SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma (MM) in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Multiple Myeloma evolves from a precursor stage called Monoclonal Gammopathy of Undetermined Significance (MGUS) to MM. Smoldering Multiple Myeloma (SMM) is an intermediate stage in this process of disease evolution.
Monoclonal gammopathies are often diagnosed by electrophoretic examination of the serum and urine proteins. They include Serum Protein ElectroPhoresis (SPEP), Serum Protein Immunofixation electrophoresis (SIFE), Urine Protein ElectroPhoresis (UPEP), Urine Protein Immunofixation electrophoresis (UIFE), and Bone Marrow evaluation. In addition, quantitative assessment of Serum Free Light Chains has been recommended for diagnosis and monitoring of neoplastic monoclonal gammopathies. 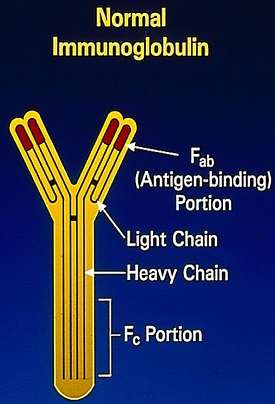
The Y-shaped Immunoglobulins are heterodimeric proteins composed of two heavy (H) and two light (L) chains. The two different types of light chains include Kappa and Lambda, which are distinctive in their amino acid sequence, with normally twice as many Kappa light chains. Malignancy can affect both types of light chains and in Multiple Myeloma. The relevant light chain production can increase, but the increase is more often in the Kappa light chains. Immunoglobulin light chains are produced in excess of the corresponding heavy chains and the excess free light chains can be quantified in serum and can also be detected in the urine, as they, by virtue of their size, are freely filtered through the glomerulus (Bence Jones protein). Excess amounts of free monoclonal light chains in patients with monoclonal gammopathy can produce nephropathy due to precipitation of these proteins in renal tubules.
Serum free Kappa and Lambda light chains are normally present in a ratio of about 0.26 to 1.65 and this ratio is increased in patients with Kappa light chain monoclonal gammopathy and decreased in patients with Lambda chain producing monoclonal gammopathies. Even though alteration in the serum K/L ratio is an important diagnostic criterion for plasma cell neoplasms, there is a high rate of positive results in patients receiving tertiary care, with abnormal K/L ratio noted in 36% of patients and about 90% of these are Kappa light chain dominant.
The serum K/L ratio is however less frequently abnormal and stays normal in patients with Lambda chain lesions even when an abnormal Lambda immunoglobulin is detected in the urine. These variabilities can result in the less-common Lambda chain-associated lesions going undiagnosed. There is a high false negative rate for Lambda dominant K/L ratio in Lambda chain associated monoclonal gammopathy (89% for MGUS, 60% for SMM and 51% for MM). It is estimated that the overall excess false negative K/L ratio rate for Lambda chain lesions, compared to Kappa chain lesions, is approximately 30%. The high false negative rate for the Lambda dominant K/L ratio, in patients with Lambda chain neoplastic monoclonal gammopathies, may be due to under-detection of Lambda light chains, Lambda chains are not produced in as much excess as are Kappa chains resulting in lower rates of Lambda dominant K/L ratio in patient with Lambda light chain neoplastic monoclonal gammopathy, and overproduction of polyclonal Kappa light chains in Lambda chain monoclonal gammopathies, as is usually noted in patients receiving tertiary care.
This study was undertaken by comparing the results of Serum and Urine Protein Electrophoreses with the results of Serum Free Light Chain Assay (SFLCA), to ascertain if the levels of overproduction of the Kappa and Lambda light chain types and their detection rates are different in patients with neoplastic monoclonal gammopathies. The authors performed a retrospective review of SPEP/SIFE, UPEP/UIFE, and SFLCA results from January 2010 through September 2017 from a total of 482 patients. Among these patients, 175 patients had a diagnosis of Neoplastic Monoclonal Gammopathy (MGUS, SMM or MM). , and evaluable results were available to address the questions of this study. Patients with Lymphomas and CLL were excluded.
The authors noted that the serum K/L ratios were appropriately abnormal more often in Kappa light chain disease. In contrast, in those with Lambda light chain disease, the K/L ratios were normal in about 25% of patients but free homogenous Lambda light chains were detectable in urine.
It was concluded that the serum Kappa/Lambda ratio in patients with Lambda light chain disease can be normal in a 25% of patients with Neoplastic Monoclonal Gammopathy and can be missed if not further evaluated with UPEP/UIFE. The authors comment that UPEP/UIFE is under- utilized and the study results question the medical necessity and clinical usefulness of the serum free light chain assay. Serum Free Light Chains in Neoplastic Monoclonal Gammopathies: Relative Under-Detection of Lambda Dominant Kappa/Lambda Ratio, and Underproduction of Free Lambda Light Chains, as Compared to Kappa Light Chains, in Patients With Neoplastic Monoclonal Gammopathies. Lee WS and Singh G. J Clin Med Res. 2018;10:562-569.