SUMMARY: The American Cancer Society estimates that in 2018, about 55,440 people will be diagnosed with pancreatic cancer and about 44,330 people will die of the disease. Pancreatic cancer is the fourth most common cause of cancer-related deaths in the United States and Western Europe. Unfortunately, unlike other malignancies, very little progress has been made and outcomes for patients with advanced pancreatic cancer, has been dismal. Diagnosis is often made late in the course of the disease, as patients are often asymptomatic and early tumors cannot be detected during routine physical examination. Further, precursors of pancreatic cancer evolve as microscopic lesions in the ducts and are often not visualized on imaging studies. Based on the National Cancer Institute Data Base, the 5 year observed survival rate for patients diagnosed with exocrine cancer of the Pancreas is 14% for those with Stage IA disease, and 1% for those with Stage IV disease. Early detection and cancer prevention is therefore critical.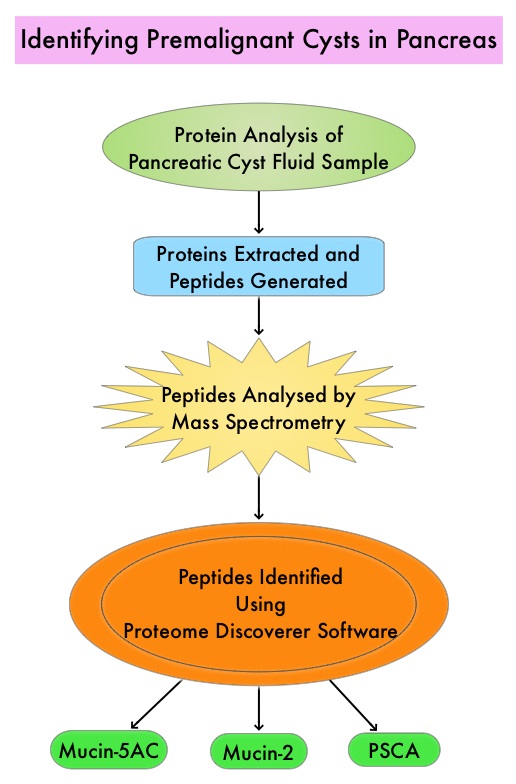
Pancreatic adenocarcinoma can also develop from mucin-producing Pancreatic Cystic Lesions (PCLs) and these neoplasms include Intraductal Papillary Mucinous Neoplasms (IPMNs) and Mucinous Cystic Neoplasms. These neoplasms comprise 10-50% of Pancreatic Cystic Lesions (PCLs). It should be noted that PCLs also encompass intrinsically benign tumors such as serous cystic neoplasms and inflammatory pseudocysts. With the rising use of abdominal MRI, partly due to concerns about ionizing radiation inherent to other exams such as CT scans, PCLs are incidentally discovered in up to 20% of these imaging studies in adults and these individuals are asymptomatic. Imaging techniques that are presently available cannot distinguish between benign, premalignant, and malignant PCLs. The same is true for currently available Endoscopic Ultrasound (EUS)-guided Fine Needle Aspiration (FNA) of Pancreatic Cystic Lesions and evaluation of cyst fluid for cytology and quantification of CarcinoEmbryonic Antigen (CEA). A high risk lesion in the pancreas would require surgical intervention with associated risks. Identifying benign from premalignant and malignant PCLs, as well as determining the epithelial subtype of IPMNs is therefore critical. The risk of malignancy is highest for Pancreatobiliary-type IPMNs with somewhat better prognosis for Intestinal-type IPMNs, whereas Gastric-type IPMNs tend to be indolent.
The authors in this study utilized targeted Mass Spectrometry (MS) to identify and quantitate proteins in the cystic fluid samples. Targeted quantitation of proteins by Mass Spectrometry provides a next-generation platform that overcomes many of the limitations of Western blotting and provides new capabilities for protein analysis. This sensitive technique is used to detect, identify and quantitate protein molecules in a given sample, based on their mass-to-charge ratio, enabling targeted protein measurement.
Using pancreatic cyst fluid samples obtained by routine EUS-guided FNA, biomarker candidates for malignant potential and high-grade dysplasia/cancer were identified via an explorative proteomic approach, in an initial cohort of 24 patients. Subsequently, a quantitative analysis using 30 heavy-labeled peptides from the biomarkers and parallel reaction monitoring mass spectrometry was devised, and tested, in a training cohort of 80 patients, and prospectively evaluated in a validation cohort of 68 patients. Patients with solid-pseudopapillary neoplasm and neuroendocrine tumor were excluded. The Primary objective of this study was to devise and validate a targeted, quantitative proteomic analysis to identify and distinguish between premalignant Pancreatic Cystic Lesions (PCLs) and Cystic neoplasms with manifest high-grade dysplasia /cancer. A Secondary aim was to find and evaluate markers for different epithelial subtypes of IPMNs, which may be used to predict the risk of malignant transformation.
It was noted that the optimal set of markers for detecting malignant potential was a panel of peptides from Mucin-5AC and Mucin-2, which could distinguish premalignant/malignant lesions from benign, with an accuracy of 97% in the validation cohort , compared with 61% using pancreatic cyst fluid CarcinoEmbryonic Antigen (P< 0.001) and 84% using Cytology (P=0.02). A combination of proteins Mucin-5AC and Prostate Stem Cell Antigen (PSCA) could identify high-grade dysplasia/cancer with an accuracy of 96% and detected 95% of malignant/severely dysplastic lesions, compared with 35% and 50% for CarcinoEmbryonic Antigen and Cytology (P<0.001 and P=0.003, respectively).
The authors concluded that Targeted Mass Spectrometry analysis of three pancreatic cyst fluid biomarkers provides highly accurate identification and assessment of cystic precursors to pancreatic adenocarcinoma. It remains to be seen whether this methodology will be beneficial for early diagnosis as well as prevention of development of pancreatic adenocarcinoma. Highly Accurate Identification of Cystic Precursor Lesions of Pancreatic Cancer Through Targeted Mass Spectrometry: A Phase IIc Diagnostic Study. Jabbar KS, Arike L, Hansson GC, et al. J Clin Oncol 2018;36:367-375