
The FDA on July 1, 2021 approved RYLAZE® as a component of a multi-agent chemotherapeutic regimen for the treatment of Acute Lymphoblastic Leukemia (ALL) and Lymphoblastic Lymphoma, in adult and pediatric patients 1 month or older who have developed hypersensitivity to E. coli-derived asparaginase. RYLAZE® is a product of Jazz Pharmaceuticals.
Author: RR
Immune Checkpoint Inhibitor Therapy and Risk of Venous Thromboembolism
SUMMARY: The Center for Disease Control and Prevention (CDC) estimates that approximately 1-2 per 1000 individuals develop Deep Vein Thrombosis (DVT)/Pulmonary Embolism (PE) each year in the United States, resulting in 60,000-100,000 deaths. Venous ThromboEmbolism (VTE) is the third leading cause of cardiovascular mortality, after myocardial infarction and stroke. Ambulatory cancer patients initiating chemotherapy are at varying risk for Venous Thromboembolism (VTE), which in turn can have a substantial effect on health care costs, with negative impact on quality of life.
Approximately 20% of cancer patients develop VTE and about 20% of all VTE cases occur in patients with cancer. Cancer patients have a 4-7 fold increased risk of thrombosis, compared with those without cancer, and patients with cancer and VTE are at a markedly increased risk for morbidity and mortality. The etiology of thrombosis in cancer is multifactorial, and the vascular system is an important interface between the malignant cells and their systemic and external environments. Genetic alterations in malignant cells, as they respond to their microenvironment, can result in inflammation, angiogenesis, and tissue repair. This in turn leads to the local and systemic activation of the coagulation system. It has been postulated that the procoagulant effect of malignant cells may be related to the release of soluble mediators such as G-CSF into the circulation or by the shedding of procoagulant Extracellular Vesicles (EVs) harboring Tissue Factor. Previously published studies had entertained the notion that certain oncogenic mutations may deregulate hemostatic genes (coagulome) in cancer cells.
Immune Checkpoint Inhibitors (ICIs) have revolutionized cancer management. They bind to either PD-1 receptor or its ligand PD-L1 and block their interaction, thereby reversing the PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. This can however be accompanied by various off-target manifestations of autoimmunity induced by immune checkpoint inhibitors with resulting systemic inflammation on the hemostatic system. The risk of Venous ThromboEmbolism (VTE) and Arterial ThromboEmbolism (ATE) associated with ICIs is currently unclear. The goal of this study was to quantify the risk of VTE/ATE in patients with cancer, treated with ICIs, explore clinical impact, and investigate potential clinical risk factors.
The authors conducted a single-center, retrospective cohort study at the Medical University of Vienna, Austria, and included 672 patients with histologically confirmed cancer, who were treated with one or more doses of an Immune Checkpoint Inhibitor (Nivolumab, Pembrolizumab, Ipilimumab, Atezolizumab, or Avelumab), between 2015 and 2018. Patients received a median of 7 cycles of therapy. About a third of patients (30.4%) had Malignant Melanoma, whereas 24% had Non Small Cell Lung Cancer, 11% had Renal Cell Carcinoma, 10.4% had Head and Neck Squamous Cell Carcinoma and 5% had Urothelial cancer. Majority of patients (86%) had advanced disease at the time of ICI initiation. The median patient age was 64 years, 39% were female and most patients had an ECOG Performance Status of 0 or 1. Approximately 13% of patients had a history of VTE prior to the initiation of ICI therapy. Approximately 9% of patients had a history of ATE, and was associated with the current cancer diagnosis in 2.2% of the total cohort. At the time of ICI therapy initiation, 16.5% received continuous anticoagulation and 20% received antiplatelet therapy. The Primary outcomes of the study were cumulative incidence rates of VTE and ATE. Secondary outcomes included the association of VTE/ATE with Overall Survival (OS), Progression Free Survival (PFS), and radiological Disease Control Rate (DCR). The median follow up was 8.5 months.
It was noted that the cumulative incidences of VTE and ATE during ICI therapy were 12.9% and 1.8% respectively. The occurrence of VTE was associated with increased mortality with shorter OS. The median OS after the occurrence of VTE was 11.6 months compared with 25.5 months in those without VTE (P<0.001). The researchers noted that the number of fatal Pulmonary Embolisms were not high (N=2) in this study, suggesting that the impact of VTE goes beyond direct VTE-related mortality. The diagnosis of VTE was further associated with shorter PFS. Median PFS after VTE was 1.7 months compared with 6.7 months in those without VTE (P<0.001). The occurrence of ATE was not associated with risk of mortality or early progression of disease. However, this could have been due to relatively low number of ATE events and potential lack of statistical power. Therefore, definite conclusions cannot be drawn. Prior history of VTE predicted VTE occurrence. Distant metastasis was associated with VTE risk, although this was not statistically significant. The researchers did not find association of VTE with ECOG Performance Status or Khorana score, and the rates of VTE were comparable between tumor types and different immune Checkpoint Inhibitors. No association with VTE risk was observed for patients undergoing continuous anticoagulation or anti-platelet therapy at baseline.
It was concluded that despite the limitations of this study, patients with cancer undergoing treatment with Immune Checkpoint inhibitors are at a high risk of developing thromboembolic complications, especially VTE, and VTE occurrence was associated with increased mortality. The authors added that further studies are needed to better understand the risk of VTE and ATE associated with ICIs, and thus improve patient care by preventing thromboembolic complications.
Incidence, risk factors, and outcomes of venous and arterial thromboembolism in immune checkpoint inhibitor therapy. Moik F, Chan WE, Wiedemann S, et al. Blood.2021;137:1669-1678.
FDA Approves LENVIMA® Plus KEYTRUDA® for Advanced Renal Cell Carcinoma
SUMMARY: The FDA on August 10, 2021, approved the combination of LENVIMA® (Lenvatinib) plus KEYTRUDA® (Pembrolizumab) for first line treatment of adult patients with advanced Renal Cell Carcinoma (RCC). The American Cancer Society estimates that 76,080 new cases of kidney cancers will be diagnosed in the United States in 2021 and about 13,780 people will die from the disease. Renal Cell Carcinoma (RCC) is by far the most common type of kidney cancer and is about twice as common in men as in women. Modifiable risk factors include smoking, obesity, workplace exposure to certain substances and high blood pressure. The five year survival of patients with advanced RCC is less than 10% and there is a significant unmet need for improved therapies for this disease.
SUTENT® (Sunitinib) is a MultiKinase Inhibitor (MKI) which simultaneously targets the tumor cell wall, vascular endothelial cell wall as well as the pericyte/fibroblast/vascular/smooth vessel cell wall, and is capable of specifically binding to tyrosine kinases inhibiting the earlier signaling events and thereby inhibits phosphorylation of VEGF receptor, PDGF receptor, FLT-3 and c-KIT. SUTENT® has been the standard first line intervention for treatment naïve patients with advanced RCC. In a large, multi-center, randomized, Phase III study, the median Progression Free Survival (PFS) with SUTENT® was 9.5 months, the Objective Response Rate (ORR) was 25%, and the median Overall Survival (OS) was 29.3 months, when compared with Interferon Alfa, in patients with treatment-naïve Renal Cell Carcinoma. This was however associated with a high rate of hematological toxicities.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells.
LENVIMA® (Lenvatinib) is an oral multitargeted TKI which targets Vascular Endothelial Growth Factor Receptor (VEGFR) 1-3, Fibroblast Growth Factor Receptor (FGFR) 1-4, Rearranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with antiangiogenesis properties by its ability to inhibit FGFR-1, thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting FGFR and PDGFR beta.
AFINITOR® (Everolimus) does not inhibit tyrosine kinases, but is a specific inhibitor of mTOR (Mammalian Target of Rapamycin), which is a serine/threonine kinase, normally activated further downstream in the signaling cascade. With the inhibition of mTOR, protein synthesis is inhibited resulting in decreased angiogenesis, cell proliferation and survival as well as decreased levels of HIF-1 alpha.
A combination of LENVIMA® plus AFINITOR® was shown to be associated with longer Progression Free Survival than AFINITOR® alone as second line treatment in advanced RCC (Lancet Oncol 2015;16:1473-1482). LENVIMA® plus KEYTRUDA® was shown to have promising antitumor activity in previously treated patients with RCC in a Phase IB-II trial (J Clin Oncol 2020;38:1154-1163). Based on this data, the authors conducted a multicenter, randomized, open-label, Phase III trial to compare the efficacy and safety of LENVIMA® in combination with KEYTRUDA® or AFINITOR® versus SUTENT® alone, in first line treatment of patients with advanced RCC.
The researchers randomly assigned 1069 patients with advanced RCC and no previous systemic therapy in a 1:1:1 ratio to receive LENVIMA® 20 mg orally once daily plus KEYTRUDA® 200 mg IV once every 3 weeks (N=355), LENVIMA® 18 mg orally once daily plus AFINITOR® 5 mg orally once daily (N=357) or SUTENT® 50 mg orally once daily, alternating 4 weeks on and 2 weeks off (N=357). The Primary end point was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR) and Safety. The median follow up for OS was 26.6 months.
The median PFS was significantly longer with LENVIMA® plus KEYTRUDA® combination, compared to single agent SUTENT® (23.9 months versus 9.2 months, HR=0.39; P<0.001). The median PFS with the LENVIMA® plus AFINITOR® combination was also significantly longer, compared to single agent SUTENT® (14.7 months versus 9.2 months, HR=0.65; P<0.001). The PFS benefit favored the two LENVIMA® combination regimens over single agent SUTENT® across all evaluated subgroups, including those based on MSKCC prognostic risk group and International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk group. At interim analysis, the OS was significantly longer with LENVIMA® plus KEYTRUDA® than with SUTENT® (HR for death=0.66; P=0.005). This benefit was noted in most subgroups, including patients with PD-L1 positive or negative tumors, with an exception of patients with favorable risk disease as defined by IMDC criteria. Overall Survival with LENVIMA® plus AFINITOR® was however not significantly longer compared with SUTENT® (HR=1.15; P=0.30).
The confirmed ORR was 71% with LENVIMA® plus KEYTRUDA®, 53.5% with LENVIMA® plus AFINITOR®, and 36.1% with single agent SUTENT®. The Complete Response rate was 16.1% in the LENVIMA® plus KEYTRUDA® group, 9.8% in the LENVIMA® plus AFINITOR® group, and 4.2% in the SUTENT® group. The median Duration of Response in patients who had a confirmed response was 25.8 months in the LENVIMA® plus KEYTRUDA® group, 16.6 months in the LENVIMA® plus AFINITOR® group, and 14.6 months in the SUTENT® group. Grade 3 or higher Adverse Events occurred in 82.4% of the patients who received LENVIMA® plus KEYTRUDA® group, in 83.1% of the patients who received LENVIMA® plus AFINITOR®, and in 71.8% of the patients who received SUTENT®.
It was concluded that a combination of LENVIMA® plus KEYTRUDA® provided superior Progression Free Survival and Overall Survival compared to SUTENT®, in the first line treatment of patients with advanced Renal Cell Carcinoma.
Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. Motzer R, Alekseev B, Rha S-Y, et al. for the CLEAR Trial Investigators. N Engl J Med 2021; 384:1289-1300
AYVAKIT® (Avapritinib)
The FDA on June 16, 2021, approved AYVAKIT® for adult patients with advanced Systemic Mastocytosis, including patients with aggressive Systemic Mastocytosis, Systemic Mastocytosis with an associated hematological neoplasm, and Mast Cell Leukemia. AYVAKIT® is a product of Blueprint Medicines Corp.
TRUSELTIQ® (Infigratinib)
The FDA on May 28, 2021, granted accelerated approval to TRUSELTIQ®, a kinase inhibitor, for adults with previously treated, unresectable locally advanced or metastatic Cholangiocarcinoma with a Fibroblast Growth Factor Receptor 2 (FGFR2) fusion or other rearrangement, as detected by an FDA-approved test. TRUSELTIQ® is a product of QED Therapeutics, Inc.
The FDA also approved FoundationOne® CDx (Foundation Medicine, Inc.) for selection of patients with FGFR2 fusion or other rearrangement as a companion diagnostic device for treatment with TRUSELTIQ®.
KRAS Variant Status and Outcomes with Immune Checkpoint Inhibitor-Based Therapy in Advanced Non Small Cell Lung Cancer
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Patients with advanced NSCLC without a driver mutation and with Programmed cell Death Ligand 1 (PD-L1) expression of 50% or greater, are often treated first line with Immune Checkpoint Inhibition (ICI) monotherapy or ICI in combination with chemotherapy. The choice between these two treatment regimens is usually based on tumor burden and patient comorbidities, as there are presently no biomarkers available to predict the risk and benefit of these treatment interventions. The KEYNOTE-042 study demonstrated that single agent Pembrolizumab given as first line therapy demonstrated Overall Survival (OS) benefit over chemotherapy, in patients with previously untreated advanced NSCLC, with PD-L1 expression of 1% or greater. In an exploratory analysis, this benefit was seen regardless of KRAS status, but was more pronounced in patients with KRAS variants than those without KRAS variants.
The KRAS (kirsten rat sarcoma viral oncogene homologue) proto-oncogene encodes a protein that is a member of the small GTPase super family. The KRAS gene provides instructions for making the KRAS protein, which is a part of a signaling pathway known as the RAS/MAPK pathway. When mutated, KRAS oncogene has the potential to change normal cells cancerous. KRAS is the most frequently mutated oncogene in human cancers and are often associated with resistance to targeted therapies and poor outcomes. The KRAS-G12C mutation occurs in approximately 12-15% of NSCLC and in 3-5% of Colorectal cancers and other solid cancers. KRAS G12C is one of the most prevalent driver mutations in NSCLC and accounts for a greater number of patients than those with ALK, ROS1, RET, and TRK 1/2/3 mutations combined. KRAS G12C cancers are genomically more heterogeneous and occur more frequently in current or former smokers, and are likely to be more complex genomically than EGFR mutant or ALK rearranged cancers.
The authors conducted this study to evaluate the association of KRAS status with outcomes following ICI monotherapy versus chemoimmunotherapy in patients with PD-L1 of 50% or greater. The researchers used the Flatiron Health database, comprising 280 cancer clinics across the US and analyzed 1127 patients with advanced non-squamous NSCLC with PD-L1 expression of 50% or greater, known KRAS variant status, and no alteration in EGFR, ALK, or ROS1, who were treated with first line ICI monotherapy or chemoimmunotherapy between January 2016 and May 2020. Of the patients analyzed, 50.8% had KRAS variant status and 49.2% had KRAS wild type status. Patients with KRAS variant status were more likely to be female (58.7% versus 47.1%; P =0.002) and had smoking history (96.4% versus 87.7%; P < .001). Other patient demographics and patient characteristics, including age, race, ethnicity, Performance Status, and stage at diagnosis, were well balanced among the groups analyzed. Patient groups were stratified by treatment type and KRAS status (variant or wild type), and Overall Survival (OS) was compared between the treatment groups. Adjusted Hazard ratios for death associated with KRAS status and treatment regimen was estimated, using Cox proportional hazards models.
It was noted that among patients treated with ICI monotherapy, KRAS variant status was associated with superior median survival compared with KRAS wild type (21.1 months versus 13.6 months; HR=0.77; P=0.03), and this was statistically significant. However, among patients treated with chemoimmunotherapy, there was no significant median survival difference between patients with KRAS variant and KRAS wild type status (20.0 months versus 19.3 months; HR=0.99; P=0.93).
Among patients with KRAS variant status, the median OS did not differ between those treated with ICI monotherapy and chemoimmunotherapy (21.1 months versus 20.0 months; P =0.78), whereas among patients with KRAS wild type status, those treated with ICI monotherapy had numerically worse median survival than those treated with chemoimmunotherapy, although this difference was not statistically significant (13.6 months versus 19.3 months; HR=1.19; P =0.06).
In conclusion, this data suggests that chemoimmunotherapy might be favored over ICI monotherapy for patients with KRAS wild type tumors associated with high PD-L1 expression. The authors caution that in this analysis KRAS variant subtype and co-mutation status including TP53 and STK11 was unknown, and further investigation is needed to selection appropriate therapies for patients with PD-L1 High NSCLC.
Association Between KRAS Variant Status and Outcomes With First-line Immune Checkpoint Inhibitor–Based Therapy in Patients With Advanced Non–Small-Cell Lung Cancer. Sun L, Hsu M, Cohen RB, et al. JAMA Oncol. 2021;7:937-939.
Long Term Results in Prostate Cancer Patients after Short Term Androgen Suppression and Radiation Dose Escalation
SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer, and 1 in 8 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 248,530 new cases of prostate cancer will be diagnosed in 2021 and 34,130 men will die of the disease.
The development and progression of prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) or testosterone suppression has therefore been the cornerstone of treatment of advanced prostate cancer, and is the first treatment intervention. Treatment options for patients with intermediate and high risk prostate cancer include Radical Prostatectomy and External Beam Radiation Therapy (EBRT), and long term outcomes are similar with both treatment approaches. For those receiving EBRT, based on several clinical trials, a minimum radiation dose of 74 Gy is recommended for both intermediate and high risk prostate cancer patients. Numerous studies have demonstrated the benefit of combining Androgen Suppression (AS) with EBRT as initial treatment of localized prostate cancer. However, the optimal Androgen Suppression treatment duration has remained unclear. For patients with intermediate risk disease, 4-6 months of neoadjuvant/adjuvant Androgen Suppression is considered sufficient, whereas 2-3 years of Androgen Suppression is recommended for localized high risk disease.
EORTC 22991 was launched in 2001 to assess the benefit of 6 months of concomitant and adjuvant Androgen Suppression added to EBRT in men with intermediate and limited high risk localized prostate cancer. Eligible patients had histologically confirmed prostate adenocarcinoma (T1b-T2b), with PSA 10-20 ng/mL and/or Gleason sum equals 7, or patients with PSA less than 10 ng/mL, Gleason sum less than 7 and cT2b disease. Patients had no involvement of pelvic lymph nodes (N0) as assessed by CT or MRI or laparoscopic surgery, no clinical evidence of metastatic spread (M0), no previous pelvic irradiation or radical prostatectomy and no previous hormonal therapy. Either, a PSA of more than 20 ng/mL, a Gleason sum more than 7, or a disease stage more than cT2c, was classified high-risk disease, and these patients were ineligible. Patients (N=819) were randomly assigned to receive EBRT or EBRT plus Androgen Suppression, started on day 1 of EBRT. The treating Radiation Therapy (RT) facilities centers selected the EBRT dose (70Gy, 74Gy, or 78 Gy) as well as RT technique (3D-Conformal Radiation Therapy or IMRT). Androgen Suppression consisted of two subcutaneous injections of 3-monthly depot LHRH analog (Goserelin) given the first day of RT and then 3 months later. To avoid Flare phenomenon, patients received antiandrogen agent, Bicalutamide 50 mg orally daily for one month, starting 1 week before the first LHRH injection. Of the 481 patients with intermediate-risk disease, 342 patients had EBRT planned at 74 Gy and 139 patients had EBRT planned at 78 Gy. Of the 481 patients, 245 were randomly assigned to EBRT plus Androgen Suppression (173 patients at 74 Gy, 72 patients at 78 Gy) and 236 patients to EBRT only (169 patients at 74 Gy, 67 patients at 78 Gy). The Primary endpoint was Event Free Survival (EFS). Secondary end points included clinical Disease Free Survival (DFS), Overall Survival (OS) and Distant Metastasis Free Survival (DMFS).
The authors in 2016 reported that after a median follow up of 7.2 years, 6-month concomitant and adjuvant AS combined with EBRT improved 5-year EFS and clinical DFS of intermediate and limited high risk prostate cancer patients, compared with those treated with EBRT alone. The researchers in this publication reported the updated results. Limited high risk patients, and all patients treated with EBRT at 70 Gy were excluded from this present analysis, because this is considered suboptimal according to the current practice standards.
At a median follow up of 12.2 years, it was confirmed that the addition of 6 months of concomitant and adjuvant Androgen Suppression significantly improved EFS (HR=0.53; P<0.001), clinical DFS (HR=0.67; P=0.008) and locoregional control (HR=0.44; P=0.013), compared to EBRT alone. These benefits were seen across age groups (less than 70 yrs versus 70 yrs or more), and were independent of radiation dose (74Gy versus 78 Gy). The observed improvements in DMFS and OS however did not reach statistical significance and this was expected, as the trial was not powered to detect difference in these endpoints.
It was concluded that External Beam Radiation Therapy at 74 or 78 Gy, along with 6 months of concomitant and adjuvant Androgen Suppression, significantly improves Event Free Survival and Disease Free Survival in intermediate risk prostate carcinoma. The authors added that these are the most robust data yet, from a randomized trial with long term follow up, addressing this important question.
Short Androgen Suppression and Radiation Dose Escalation in Prostate Cancer: 12-Year Results of EORTC Trial 22991 in Patients With Localized Intermediate-Risk Disease. Bolla M, Neven A, Maingon P, et al. DOI: 10.1200/JCO.21.00855 Journal of Clinical Oncology. Published online July 26, 2021.
Duration of Adjuvant Aromatase Inhibitor Therapy in Postmenopausal Breast Cancer
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 284,200 new cases of breast cancer will be diagnosed in 2021 and about 44,130 individuals will die of the disease, largely due to metastatic recurrence.
Luminal breast cancer is the most prevalent molecular subtype in postmenopausal females, accounting for over 70%. Despite substantial improvements in adjuvant therapies, the risk of disease recurrence continues indefinitely, with more than half the recurrences occurring after the first 5 years following diagnosis. Following initial adjuvant endocrine therapy with Tamoxifen for 5 years, the addition of extended adjuvant therapy has resulted in 40% longer Disease Free Survival (DFS), when compared to placebo or no extended therapy. However the benefit of extending adjuvant Aromatase Inhibitor therapy for 5 years beyond the initial 5-year duration regimen is less well established. Further, the most effective duration of such extended adjuvant endocrine therapy remains unclear. Added to this dilemma are the side effects associated with Aromatase Inhibitor therapy including hot flushes, arthralgia, and bone pain, as well as treatment-induced osteoporosis, which can have a significant impact on patient’s quality of life. Researchers in the Secondary Adjuvant Long-Term Study with Arimidex [Anastrozole] (SALSA) prospectively investigated whether an additional 2 years or 5 years of Anastrozole therapy would result in better outcomes, following the initial 5 years of endocrine therapy, in postmenopausal women with Hormone Receptor-positive breast cancer.
The authors conducted a prospective, multicenter, randomized, Phase III trial, which included 3,470 eligible postmenopausal women with Stages I, II or III early stage breast cancer with no evidence of recurrence. Enrolled patients had invasive Hormone Receptor-positive breast cancer, and had received 5 years (plus or minus 12 months) of adjuvant endocrine therapy with Tamoxifen, Aromatase Inhibitors, or both sequentially, up until 12 months before randomization. Patients were randomly assigned 1:1 to receive Anastrozole 1 mg, orally daily, for either 2 additional years for a total of 7 years (N=1732) or 5 additional years for a total of 10 years (N=1738). The two treatment groups were well balanced. The median age at the time of randomization was 64 years, 72% of patients had tumors that were smaller than 2 cm, 66% had node-negative disease, and 19% had high-grade tumors. Stratification criteria included pathological tumor stage, pathological node stage, primary adjuvant endocrine therapy and adjuvant chemotherapy.
The primary analysis included all the patients who were still participating in the study (N=3208), including 1,603 in the 2-year group versus 1,605 in the 5-year group. In the primary analysis population of 3208 patients, 51% had received Tamoxifen alone for the initial 5 years, 7.3% had received an Aromatase Inhibitor alone, and 41.7% had received an Aromatase Inhibitor in combination with Tamoxifen. The Primary end point was Disease Free Survival (DFS). Secondary end points were Overall Survival (OS), contralateral breast cancer, second primary cancer, and clinical bone fracture. The median follow-up after randomization was 118 months.
The researchers observed no difference in DFS with 2 versus 5 additional years of adjuvant endocrine therapy with Anastrozole. The DFS 10 years since randomization was 73.6% in the 2-year group versus 73.9% in the 5-year group (HR=0.99; P=0.90). Contralateral breast cancer occurred in 2.2% versus 2.1% of patients (HR= 1.15), and local recurrence occurred in 3% versus 2.4% in the 2 year and 5 year groups, respectively. There was no difference noted for Overall Survival at 8 years between the two treatment groups (87.5% in the 2-year group and 87.3% in the 5-year group, HR for death from any cause=1.02). The risk of clinical bone fracture however was higher in the 5-year group than in the 2-year group (HR=1.35).
It was concluded from this study that in postmenopausal women with Hormone Receptor positive breast cancer who had received 5 years of adjuvant endocrine therapy, extending endocrine therapy with an Aromatase Inhibitor by an additional 5 years provided no benefit over a 2-year extension, but was associated with a greater risk of bone fracture.
Duration of Adjuvant Aromatase-Inhibitor Therapy in Postmenopausal Breast Cancer. Gnant M, Fitzal F, Rinnerthaler G, et al. N Engl J Med 2021; 385:395-405.
CALQUENCE® May Be Safer Than IMBRUVICA® in CLL Patients
SUMMARY: The American Cancer Society estimates that for 2021, about 21,250 new cases of Chronic Lymphocytic Leukemia (CLL) will be diagnosed in the US and 4320 patients will die of the disease. CLL accounts for about one-quarter of the new cases of leukemia. The average age of patients diagnosed with CLL is around 70 years, and is rarely seen in people under age 40, and is extremely rare in children.
Bruton’s Tyrosine Kinase (BTK) is a member of the Tec family of kinases, downstream of the B-cell receptor and is predominantly expressed in B-cells. It is a mediator of B-cell receptor signaling in normal and transformed B-cells. Ibrutinib (IMBRUVICA®) is an oral, irreversible inhibitor of BTK and inhibits cell proliferation and promotes programmed cell death (Apoptosis) by blocking B-cell activation and signaling. Ibrutinib demonstrated survival benefits when compared to chemoimmunotherapy both in previously untreated (RESONATE-2), as well as relapsed (RESONATE) CLL patients. However, toxicities leading to Ibrutinib discontinuation occurred in a significant number of patients, and Atrial Fibrillation was noted in 11-16% of patients and hypertension rates were between 20-26%.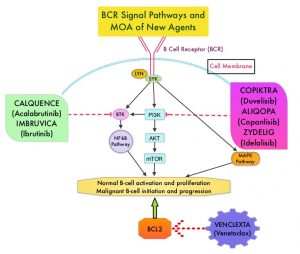
Acalabrutinib (CALQUENCE®) is a highly selective, next-generation, oral, covalent, irreversible Bruton Tyrosine Kinase (BTK) inhibitor with minimal activity against other kinases. Acalabrutinib has a shorter plasma half-life and inhibits cell proliferation and promotes programmed cell death (Apoptosis) by blocking B-cell activation and signaling. Acalabrutinib demonstrated superior Progression Free Survival (PFS) versus chemoimmunotherapy in patients with previously untreated (ELEVATE-TN), as well as Relapsed or Refractory (ASCEND) CLL. Acalabrutinib was better tolerated with lower rates of treatment discontinuation due to adverse events, and also demonstrated efficacy and tolerability in Ibrutinib-intolerant patients with CLL.
ELEVATE-RR is a prospective, randomized, multicenter, open-label, noninferiority, Phase III study, conducted to compare the efficacy and safety of Acalabrutinib with Ibrutinib, in patients with previously treated CLL, and to test the hypothesis that Acalabrutinib was noninferior to Ibrutinib in PFS, with improved tolerability. This trial included 533 previously treated patients with CLL who were randomly assigned 1:1 to receive Acalabrutinib 100 mg orally twice daily (N=268) or Ibrutinib 420 mg orally once daily (N=265). Treatment was continued until disease progression or unacceptable toxicity, and crossover between treatment groups was not permitted. Enrolled patients had centrally confirmed del(17)(p13.1) or del(11)(q22.3) and patients were stratified based on cytogenetics, ECOG Performance Status and number of prior therapies. Patients with significant cardiovascular disease, concomitant vitamin K antagonist treatment, prior BTK or BCL2 inhibitor treatment, or those requiring treatment with Proton-Pump Inhibitors were excluded. The Primary end point was Independent Review Committee-assessed noninferiority of Progression Free Survival (PFS). Secondary end points included incidences of any grade Atrial Fibrillation, Grade 3 or higher infections, Richter transformation and Overall Survival (OS)
After a median follow-up of 40.9 months, Acalabrutinib was determined to be noninferior to Ibrutinib with a median PFS of 38.4 months in both arms (HR=1.00), thus meeting the noninferiority criterion. Any-grade Atrial Fibrillation/Atrial Flutter incidence was significantly lower with Acalabrutinib compared to Ibrutinib (9.4% versus 16.0%; P=0.02). Bleeding events were less frequent with Acalabrutinib (38%) versus Ibrutinib (51.3%). The median Overall Survival was not reached in either treatment groups. Treatment was discontinued due to adverse events in 14.7% of Acalabrutinib-treated patients and 21.3% of Ibrutinib-treated patients.
The authors concluded that this is the first direct comparison of Ibrutinib with Acalabrutinib in CLL and Acalabrutinib demonstrated noninferior PFS and provides improved safety, with fewer Atrial Fibrillation events and discontinuations because of adverse events, when compared to Ibrutinib.
Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. Byrd JC, Hillmen P, Ghia P, et al. DOI: 10.1200/JCO.21.01210 Journal of Clinical Oncology. Published online July 26, 2021.
FDA Approves KEYTRUDA® for High Risk Early Stage Triple Negative Breast Cancer
SUMMARY: The FDA on July 26, 2021, approved KEYTRUDA® (Pembrolizumab) for high risk, early stage, Triple Negative Breast Cancer (TNBC), in combination with chemotherapy, as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment following surgery. The FDA also granted regular approval to KEYTRUDA® in combination with chemotherapy for patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (Combined Positive Score – CPS 10 or more), as determined by an FDA approved test. FDA granted accelerated approval to KEYTRUDA® for this indication in November 2020.
Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 284,200 new cases of breast cancer will be diagnosed in 2021 and about 44,130 individuals will die of the disease, largely due to metastatic recurrence. Triple Negative Breast Cancer (TNBC) is a heterogeneous, molecularly diverse group of breast cancers and are ER (Estrogen Receptor), PR (Progesterone Receptor) and HER2 (Human Epidermal Growth Factor Receptor-2) negative. TNBC accounts for 15-20% of invasive breast cancers, with a higher incidence noted in young patients. It is usually aggressive, and tumors tend to be high grade and patients with TNBC are at a higher risk of both local and distant recurrence. Those with metastatic disease have one of the worst prognoses of all cancers with a median Overall Survival of 13 months. The majority of patients with TNBC who develop metastatic disease do so within the first 3 years after diagnosis, whereas those without recurrence during this period of time have survival rates similar to those with ER-positive breast cancers.
The lack of known recurrent oncogenic drivers in patients with metastatic TNBC, presents a major therapeutic challenge. Nonetheless, patients with TNBC often receive chemotherapy in the neoadjuvant, adjuvant or metastatic settings and approximately 30-40% of patients achieve a pathological Complete Response (pCR) in the neoadjuvant setting. In addition to increasing the likelihood of tumor resectability and breast preservation, patients achieving a pCR following neoadjuvant chemotherapy have a longer Event Free Survival (EFS) and Overall Survival (OS). Those who do not achieve a pathological Complete Response tend to have a poor prognosis. For all these reasons, pCR is considered a valid endpoint for clinical testing of neoadjuvant therapy in patients with early stage TNBC. It appears that there are subsets of patients with TNBC who may be inherently insensitive to cytotoxic chemotherapy. Three treatment approaches appear to be promising and they include immune therapies, PARP inhibition and inhibition of PI3K pathway. Previously published studies have shown that presence of tumor-infiltrating lymphocytes was associated with clinical benefit, when treated with chemotherapy and immunotherapy, in patients with TNBC, and improved clinical benefit was observed in patients with immune-enriched molecular subtypes of metastatic TNBC.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. Cytotoxic chemotherapy releases tumor-specific antigens and immune checkpoint inhibitors such as KEYTRUDA® when given along with chemotherapy can enhance endogenous anticancer immunity. Preliminary results from Phase I and II trials have shown that in patients with TNBC, KEYTRUDA® given along with chemotherapy in a neoadjuvant setting resulted in a high rate of pCR.
The present FDA approvals were based on KEYNOTE-522, which is an international, randomized, multicenter, double-blind, placebo controlled Phase III trial, conducted to evaluate the safety and efficacy of neoadjuvant KEYTRUDA® plus chemotherapy followed by adjuvant KEYTRUDA® or placebo, in patients with early stage TNBC. In this study, 1,174 patients were randomly assigned in a 2:1 ratio to receive neoadjuvant KEYTRUDA® 200 mg IV every 3 weeks (N=784) or placebo (N=390). All patients received 4 cycles of Carboplatin plus Paclitaxel, followed by 4 cycles of Doxorubicin or Epirubicin plus Cyclophosphamide, in the neoadjuvant setting. Following definitive surgery, adjuvant KEYTRUDA® or placebo was continued every 3 weeks for 9 cycles or until disease recurrence or unacceptable toxicity. Enrolled TNBC patients were newly diagnosed, early stage, high risk, treatment naïve, and included both node-negative and node-positive patients with nonmetastatic disease (Tumor Stage T1c, Nodal Stage N1-N2 or Tumor Stage T2-T4, Nodal Stage N0-N2, per AJCC criteria). Patients were enrolled regardless of tumor PD-L1 expression. Treatment groups were well balanced and patients were stratified according to nodal status, tumor size, and Carboplatin schedule (weekly versus every 3 weeks). The two Primary endpoints were pathological Complete Response (pCR) at the time of definitive surgery and Event Free Survival (EFS).
At the first interim analysis, at a median follow up of 15.5 months, the pCR among the first 602 patients who underwent randomization was 64.8% in the KEYTRUDA® plus chemotherapy group, compared with 51.2% in the placebo plus chemotherapy group (HR=0.63; P<0.001). At the median follow-up of 39 months, EFS data were made available, and this showed that KEYTRUDA® demonstrated a statistically significant EFS benefit compared with chemotherapy alone. The number of patients who experienced an EFS event was 16% and 24%, respectively (HR=0.63; P=0.00031). Among patients who were in the PD-L1 positive, defined as those with a CPS of 1 or higher, there was a 33% reduced risk of EFS events with KEYTRUDA® compared with the placebo group (HR=0.67). In the PD-L1 negative group, patients receiving the KEYTRUDA® combination had a reduced risk for EFS events by 52% compared with the placebo-chemotherapy group (HR=0.48). Across all treatment phases, Grade 3 or higher treatment-related toxicities were 78.0% in the KEYTRUDA® plus chemotherapy group and 73.0% in the placebo plus chemotherapy group
It can be concluded from this study that among patients with early stage Triple Negative Breast Cancer, the addition of KEYTRUDA® to neoadjuvant chemotherapy significantly increased the pathological Complete Response rate, compared to those who received placebo plus neoadjuvant chemotherapy, with a statistically significant Event Free Survival benefit. This KEYTRUDA® combination therapy is a meaningful milestone for breast cancer patients, and is the first immunotherapy regimen to be approved in high risk, early stage Triple Negative Breast Cancer.
https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-high-risk-early-stage-triple-negative-breast-cancer.