Written by Dr. Irfan A. Mirza
This article is sponsored and developed by Boehringer Ingelheim Pharmaceuticals
Significant strides have been made in the last decade for systemic treatment options for stage IV non-small cell lung cancer (NSCLC), including those tailored for squamous and non-squamous histology.1,2 While non-squamous NSCLC has benefited from advances such as the introduction of personalized, genotyped-directed therapies, and immunotherapy drugs, the treatment options for squamous cell NSCLC remain limited.1,2
Historically, the NCCN guidelines recommended the use of platinum-based chemotherapy in the first line setting, followed by immunotherapy in the second-line.3 However, following the results of the KEYNOTE-407 study, immunotherapy together with platinum doublet chemotherapy is now recommended in the first-line setting.4,5 This leaves an unmet need for patients with metastatic squamous NSCLC who have progressed, where most treatments consist of chemotherapy.2,6
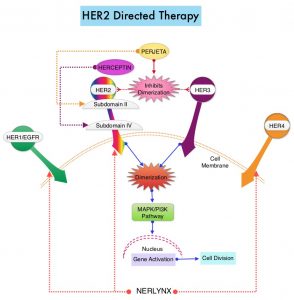
Afatinib is an oral, non-chemotherapy option for patients with metastatic squamous NSCLC who have progressed on platinum-based chemotherapy.7 Afatinib is an irreversible second-generation epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitor that selectively inhibits homo- and hetero-dimers of the ErbB receptor family (EGFR, ErbB2, and ErbB4).7
LUX-Lung 8 was a multicenter, open label, phase 3, randomized, controlled trial across 23 countries that enrolled 795 patients with advanced (stage III B and stage IV) squamous NSCLC, progressing after at least 4 cycles of platinum-based chemotherapy.8 Patients were randomized (1:1) to either afatinib 40 mg daily or erlotinib 150 mg daily until disease progression.8 The primary endpoint was progression-free survival (PFS) as assessed by an independent review committee (IRC), using RECIST v1.1 and secondary endpoints included overall survival (OS) and objective response rates as assessed by an IRC.8
In LUX-Lung 8, significant improvement in PFS and overall survival was observed for afatinib compared with erlotinib.8 The median PFS was reported as 2.4 months with afatinib and 1.9 months with erlotinib [HR, 0.82 (95% CI 0.68-0.99)] (Figure 1).8

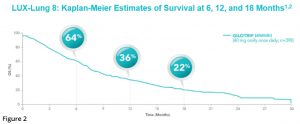
After a median follow up of 18.4 months, median OS was 7.9 months in the afatinib group and 6.8 months in the erlotinib group [HR 0.81 (95% CI 0.69-0.95), p = 0.0077].8 Estimates of OS among patients treated with afatinib were 64% at 6 months, 36% at 1 year, and 22% at 18 months (Figure 2).8
More than half (51%) of patients treated with afatinib were able to achieve disease control (defined as complete response, partial response, stable disease, or non-complete response and non-progressive disease) compared with 40% with erlotinib.8 Excluding patients with non-complete response and non-progressive disease, disease control with afatinib was 37%, vs 29% with erlotinib, in a post hoc analysis.8 The median duration of objective response was 7.3 months with afatinib and 3.7 months with erlotinib.8
The most common adverse effects associated with afatinib were diarrhea, rash/acneiform dermatitis, stomatitis, decreased appetite, nausea, vomiting, paronychia, and pruritus.8,9 Twenty percent of patients discontinued afatinib treatment due to adverse reactions, with the most frequent adverse reactions leading to discontinuation being diarrhea in 4.1% of patients and rash/acne in 2.6%.9 Serious adverse reactions occurred in 44% of patients, with pneumonia (6.6%), diarrhea (4.6%), dehydration, and dyspnea (3.1% each) being the most frequent.9 Fatal adverse reactions in afatinib-treated patients included interstitial lung disease, pneumonia, respiratory failure, acute renal failure, and general physical health deterioration, all occurring in less than 1% of patients.9
Adverse Reactions (ARs) Reported in ≥10% of GILOTRIF-Treated Patients in LUX-Lung 89*:
GILOTRIF (n=392), erlotinib (n=395) – All Grades & Grades 3-4 ARs
Gastrointestinal Disorders
Diarrhea – GILOTRIF all grades: 75%; grades 3-4: 11%; erlotinib all grades: 41%, grades 3-4: 3%
Stomatitis† – GILOTRIF all grades: 30%; grades 3-4: 4%; erlotinib all grades: 11%, grades 3-4: 1%
Nausea – GILOTRIF all grades: 21%; grades 3-4: 2%; erlotinib all grades: 16%, grades 3-4: 1%
Vomiting – GILOTRIF all grades: 13%; grades 3-4: 1%; erlotinib all grades: 10%, grades 3-4: 1%
Skin and Subcutaneous tissue disorders
Rash/acneform dermatitis‡ – GILOTRIF all grades: 70%; grades 3-4: 7%; erlotinib all grades: 70%, grades 3-4: 11%
Pruritus – GILOTRIF all grades: 10%; grades 3-4: 0%; erlotinib all grades: 13%, grades 3-4: 0%
Metabolism and nutrition disorders
Decreased appetite – GILOTRIF all grades: 25%; grades 3-4: 3%; erlotinib all grades: 13%, grades 3-4: 0%
Infections
Paronychia§ – GILOTRIF all grades: 11%; grades 3-4: 1%; erlotinib all grades: 5%, grades 3-4: 0%
*NCI CTCAE v 3.0
† Includes stomatitis, aphthous stomatitis, mucosal inflammation, mouth ulceration, oral mucosa erosion, mucosal erosion, mucosal ulceration
‡ Includes acne, dermatitis, acneiform dermatitis, eczema, erythema, exfoliative rash, folliculitis, rash, rash generalized, rash macular, rash maculo-papular,
rash pruritic, rash pustular, skin exfoliation, skin fissures, skin lesion, skin reaction, skin toxicity, skin ulcer
§ Includes paronychia, nail infection, nail bed infection
In summary, LUX-Lung 8 met its primary and secondary endpoints and remains the largest prospective head-to-head trial that compares two TKIs for second-line treatment of patients with squamous NSCLC.8 Future studies should focus on understanding the clinical profile of afatinib within the context of other commonly-used treatment modalities, such as chemotherapy. In a disease setting with few treatment options, and a pandemic which can make delivery of infusions challenging, afatinib offers patients with metastatic squamous NSCLC an opportunity to receive a chemotherapy-free, oral option once they have progressed following treatment with standard, platinum based, first line treatment.8,9
INDICATIONS AND USAGE
GILOTRIF is indicated for the treatment of patients with metastatic squamous NSCLC progressing after platinum-based chemotherapy.
IMPORTANT SAFETY INFORMATION FOR GILOTRIF® (afatinib) TABLETS
WARNINGS AND PRECAUTIONS
Diarrhea
• GILOTRIF can cause diarrhea which may be severe and can result in dehydration with or without renal impairment. In clinical studies, some of these cases were fatal.
• For patients who develop Grade 2 diarrhea lasting more than 48 hours or Grade 3 or greater diarrhea, withhold GILOTRIF until diarrhea resolves to Grade 1 or less, and then resume at a reduced dose.
• Provide patients with an anti-diarrheal agent (e.g., loperamide) for self-administration at the onset of diarrhea and instruct patients to continue anti-diarrheal until loose stools cease for 12 hours.
Bullous and Exfoliative Skin Disorders
• GILOTRIF can result in cutaneous reactions consisting of rash, erythema, and acneiform rash. In addition, palmar-plantar erythrodysesthesia syndrome was observed in clinical trials in patients taking GILOTRIF.
• Discontinue GILOTRIF in patients who develop life-threatening bullous, blistering, or exfoliating skin lesions. For patients who develop Grade 2 cutaneous adverse reactions lasting more than 7 days, intolerable Grade 2, or Grade 3 cutaneous reactions, withhold GILOTRIF. When the adverse reaction resolves to Grade 1 or less, resume GILOTRIF with appropriate dose reduction.
• Postmarketing cases of toxic epidermal necrolysis (TEN) and Stevens Johnson syndrome (SJS) have been reported in patients receiving GILOTRIF. Discontinue GILOTRIF if TEN or SJS is suspected.
Interstitial Lung Disease
• Interstitial Lung Disease (ILD) or ILD-like adverse reactions (e.g., lung infiltration, pneumonitis, acute respiratory distress syndrome, or alveolitis allergic) occurred in patients receiving GILOTRIF in clinical trials. In some cases, ILD was fatal. The incidence of ILD appeared to be higher in Asian patients as compared to white patients.
• Withhold GILOTRIF during evaluation of patients with suspected ILD, and discontinue GILOTRIF in patients with confirmed ILD.
Hepatic Toxicity
• Hepatic toxicity as evidenced by liver function tests abnormalities has been observed in patients taking GILOTRIF. In 4257 patients who received GILOTRIF across clinical trials, 9.7% had liver test abnormalities, of which 0.2% were fatal.
• Obtain periodic liver testing in patients during treatment with GILOTRIF. Withhold GILOTRIF in patients who develop worsening of liver function. Discontinue treatment in patients who develop severe hepatic impairment while taking GILOTRIF.
Gastrointestinal Perforation
• Gastrointestinal (GI) perforation, including fatal cases, has occurred with GILOTRIF. GI perforation has been reported in 0.2% of patients treated with GILOTRIF among 3213 patients across 17 randomized controlled clinical trials.
• Patients receiving concomitant corticosteroids, nonsteroidal anti-inflammatory drugs (NSAIDs), or anti-angiogenic agents, or patients with increasing age or who have an underlying history of GI ulceration, underlying diverticular disease, or bowel metastases may be at an increased risk of perforation.
• Permanently discontinue GILOTRIF in patients who develop GI perforation.
Keratitis
• Keratitis has been reported in patients taking GILOTRIF.
• Withhold GILOTRIF during evaluation of patients with suspected keratitis. If diagnosis of ulcerative keratitis is confirmed, interrupt or discontinue GILOTRIF. If keratitis is diagnosed, the benefits and risks of continuing treatment should be carefully considered. GILOTRIF should be used with caution in patients with a history of keratitis, ulcerative keratitis, or severe dry eye. Contact lens use is also a risk factor for keratitis and ulceration.
Embryo-Fetal Toxicity
• GILOTRIF can cause fetal harm when administered to a pregnant woman. Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
• Advise females of reproductive potential to use effective contraception during treatment, and for at least 2 weeks after the last dose of GILOTRIF. Advise female patients to contact their healthcare provider with a known or suspected pregnancy.
ADVERSE REACTIONS
Adverse Reactions observed in clinical trials were as follows:
Previously Treated, Metastatic Squamous NSCLC
• In GILOTRIF-treated patients (n=392) the most common adverse reactions (≥20% all grades & vs erlotinib-treated patients (n=395)) were diarrhea (75% vs 41%), rash/acneiform dermatitis (70% vs 70%), stomatitis (30% vs 11%), decreased appetite (25% vs 26%), and nausea (21% vs 16%).
• Serious adverse reactions were reported in 44% of patients treated with GILOTRIF. The most frequent serious adverse reactions reported in patients treated with GILOTRIF were pneumonia (6.6%), diarrhea (4.6%), and dehydration and dyspnea (3.1% each). Fatal adverse reactions in GILOTRIF-treated patients included ILD (0.5%), pneumonia (0.3%), respiratory failure (0.3%), acute renal failure (0.3%), and general physical health deterioration (0.3%).
DRUG INTERACTIONS
Effect of P-glycoprotein (P-gp) Inhibitors and Inducers
• Concomitant use of P-gp inhibitors (including but not limited to ritonavir, cyclosporine A, ketoconazole, itraconazole, erythromycin, verapamil, quinidine, tacrolimus, nelfinavir, saquinavir, and amiodarone) with GILOTRIF can increase exposure to afatinib.
• Concomitant use of P-gp inducers (including but not limited to rifampicin, carbamazepine, phenytoin, phenobarbital, and St. John’s wort) with GILOTRIF can decrease exposure to afatinib.
USE IN SPECIFIC POPULATIONS
Lactation
• Because of the potential for serious adverse reactions in breastfed infants from GILOTRIF, advise women not to breastfeed during treatment with GILOTRIF and for 2 weeks after the final dose.
Females and Males of Reproductive Potential
• GILOTRIF may reduce fertility in females and males of reproductive potential. It is not known if the effects on fertility are reversible.
Renal Impairment
• Patients with severe renal impairment (estimated glomerular filtration rate [eGFR] 15 to 29 mL/min/1.73 m2) have a higher exposure to afatinib than patients with normal renal function. Administer GILOTRIF at a starting dose of 30 mg once daily in patients with severe renal impairment. GILOTRIF has not been studied in patients with eGFR <15 mL/min/1.73 m2 or who are on dialysis.
Hepatic Impairment
• GILOTRIF has not been studied in patients with severe (Child Pugh C) hepatic impairment. Closely monitor patients with severe hepatic impairment and adjust GILOTRIF dose if not tolerated.
REFERENCES
1. Baxevanos P, Mountzios G. Novel chemotherapy regimens for advanced lung cancer: have we reached a plateau? Ann Transl Med. 2018;6(8):139.
2. Santos ES, Hart L. Advanced Squamous Cell Carcinoma of the Lung: Current Treatment Approaches and the Role of Afatinib. Onco Targets Ther. 2020 Sep 22;13:9305-9321.
3. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer. V.1.2016. ©National Comprehensive Cancer Network, Inc. 2016. All rights reserved. Accessed November 2, 2020. To view the most recent and complete version of the guidelines, go online to NCCN.org.
4. Paz-Ares L, et al. Pembrolizumab plus Chemotherapy for Squamous NSCLC. N Engl J Med. 2018;379: 2040-2051; DOI:10.1056/NEJMoa1810865
5. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer. V.8.2020. ©National Comprehensive Cancer Network, Inc. 2020. All rights reserved. Accessed November 2, 2020. To view the most recent and complete version of the guidelines, go online to NCCN.org.
6. Paik PK, et al. New treatment options in advanced squamous cell lung cancer. Am Soc Clin Oncol Educ Book. 2019;39:e198-e206.
7. Hirsh V. Next-Generation Covalent Irreversible Kinase Inhibitors in NSCLC: Focus on Afatinib. BioDrugs. 2015;29(3):167 183.
8. Soria JC, et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015;16(8):897 907.
9. GILOTRIF [prescribing information]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc.
Please review the Full Prescribing Information and Patient Information.
Full Prescribing Information URL: https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Gilotrif/Gilotrif.pdf?DMW_FORMAT=pdf
Patient Information URL: https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Gilotrif/Patient%20Info/gilotrif_patient%20information.pdf?DMW_FORMAT=pdf