SUMMARY: The American Cancer Society estimates that in the United States for 2020, about 8,480 new cases of Hodgkin Lymphoma will be diagnosed and about 970 patients will die of the disease. Hodgkin Lymphoma is classified into two main groups – Classical Hodgkin Lymphomas and Nodular Lymphocyte Predominant type, by the World Health Organization. The Classical Hodgkin Lymphomas include Nodular sclerosing, Mixed cellularity, Lymphocyte rich, Lymphocyte depleted subtypes and accounts for approximately 10% of all malignant lymphomas. Nodular sclerosis Hodgkin Lymphoma histology, accounts for approximately 80% of Hodgkin lymphoma cases in older children and adolescents in the United States. Classical Hodgkin Lymphoma is a malignancy of primarily B lymphocytes and is characterized by the presence of large mononucleated Hodgkin (H) and giant multinucleated Reed-Sternberg (RS) cells, collectively known as Hodgkin and Reed-Sternberg cells (HRS).The HRS cells in turn recruit an abundance of ineffective inflammatory cells and infiltrates of immune cells.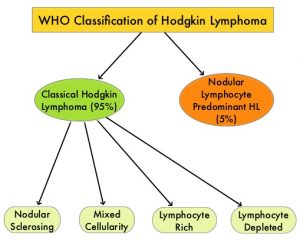
Preclinical studies suggest that HRS cells evade immune detection by exploiting the pathways associated with immune checkpoint, Programmed Death-1 (PD-1) and its ligands PD-L. Classical Hodgkin Lymphoma is an excellent example of how the tumor microenvironment influences cancer cells to proliferate and survive. The most common genetic abnormality in Nodular sclerosis subtype of Hodgkin lymphoma is the selective amplification of genes on the short arm of chromosome 9 (9p24.1) which includes JAK-2, with resulting increased expression of PD-1 ligands such as PDL1 and PDL2 on HRS cells, as well as increased JAK-STAT activity, essential for the proliferation and survival of Hodgkin Reed-Sternberg (HRS) cells. Infection with Epstein–Barr virus (EBV) similarly can increase the expression of PDL1 and PDL2 in EBV-positive Hodgkin lymphomas. It would therefore seem logical to block or inhibit immune check point PD-1 rather than both its ligands, PDL1 and PDL2.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. ADCETRIS® (Brentuximab vedotin) is an Antibody-Drug Conjugate (ADC) that targets CD30, which is a surface antigen, expressed on Reed-Sternberg cells, in patients with Classical Hodgkin Lymphoma. This ADC consists of an anti-CD30 monoclonal antibody linked to MonoMethyl Auristatin E (MMAE), an antimicrotubule agent. Upon binding to the CD30 molecule on the cancer cells, MMAE is released into the cancer cell, resulting in cell death.
Patients with Relapsed or Refractory Classical Hodgkin Lymphoma (R/R cHL) are often treated with salvage chemotherapy and Autologous Stem Cell Transplant (ASCT). There are however no standard interventions for patients ineligible for ASCT due to chemo-refractory disease, comorbidity, or advanced stage. PD-1 inhibitor such as KEYTRUDA® as well as ADCETRIS® has shown antitumor activity in R/R cHL.
KEYNOTE-204 is a randomized, international, open-label, Phase III study in which KEYTRUDA® was compared with ADCETRIS® among patients with Relapsed or Refractory Classical Hodgkin Lymphoma (R/R cHL). In this study, 304 patients were randomized 1:1, and 300 patients were treated and assigned to receive KEYTRUDA® 200 mg IV every 3 weeks (N=148) or ADCETRIS® 1.8 mg/kg IV every 3 weeks (N=152). Enrolled patients were post-Autologous Stem Cell Transplant (ASCT) or ineligible for ASCT, had measurable disease and had an ECOG Performance Status of 0 or 1. Both ADCETRIS®-naive and ADCETRIS®-exposed patients were eligible. Patients were stratified by prior ASCT and status after first-line therapy (primary refractory versus relapsed less than 12 months versus relapsed 12 months or more after end of first-line therapy). The Primary endpoints were Progression Free Survival (PFS) per Blinded Independent Central Review (BICR) and Overall Survival (OS). Secondary endpoints included PFS per investigator review, Objective Response Rate (ORR), and Safety. Median follow up was 24.7 months.
The median PFS was 13.2 months in the KEYTRUDA® group compared with 8.3 months in the ADCETRIS® group (HR=0.65, P=0.00271), suggesting an increase in PFS of 4.9 months with KEYTRUDA®. This benefit with KEYTRUDA® was observed in all subgroups tested, including those ineligible for ASCT (HR=0.61), those with primary refractory disease (HR=0.52), those who were ADCETRIS® naïve (HR=0.67), as well as those who received prior treatment with ADCETRIS® (HR=0.34). The ORR was 65.6% versus 54.2%, and the median Duration of Response was 20.7 months versus 13.8 months, in the KEYTRUDA® and ADCETRIS® groups respectively. Treatment Related Adverse Events were similar in both treatment groups and Grade 3-5 toxicities occurred in 19.6% of patients treated with KEYTRUDA® and 25% of patients treated with ADCETRIS®.
It was concluded that among patients with Relapsed/Refractory Classical Hodgkin Lymphoma, KEYTRUDA® was superior to ADCETRIS®, with a statistically significant and clinically meaningful improvement in PFS across all subgroups tested, and with safety consistent with previous reports. The authors added that KEYTRUDA® should be considered the preferred treatment option and the new standard of care in this patient population.
KEYNOTE-204: Randomized, open-label, phase III study of pembrolizumab (pembro) versus brentuximab vedotin (BV) in relapsed or refractory classic Hodgkin lymphoma (R/R cHL). Kuruvilla J, Ramchandren R, Santoro A, et al. Presented at: 2020 ASCO Virtual Scientific Program; May 29, 2020. J Clin Oncol 38: 2020 (suppl; abstr 8005).