SUMMARY: The FDA on May 1, 2020 approved DARZALEX® (Daratumumab) and Hyaluronidase-fihj (DARZALEX FASPRO®), for adult patients with newly diagnosed or Relapsed/Refractory multiple myeloma. This new product allows for subcutaneous dosing of DARZALEX®.
DARZALEX FASPRO® is now approved for these previously approved indications for IV DARZALEX®
1) In combination with VELCADE® (Bortezomib), Melphalan and Prednisone in newly diagnosed patients who are ineligible for Autologous Stem Cell Transplant (ASCT)
2) In combination with REVLIMID® (Lenalidomide) and Dexamethasone in newly diagnosed patients, who are ineligible for ASCT and in patients with Relapsed or Refractory multiple myeloma who have received at least one prior therapy
3) In combination with VELCADE® and Dexamethasone in patients who have received at least one prior therapy
4) As monotherapy, in patients who have received at least three prior lines of therapy including a Proteasome Inhibitor (PI) and an Immunomodulatory agent or who are double-refractory to a PI and an immunomodulatory agent.
DARZALEX® is a human IgG1 antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Dependent Cytotoxicity (CDC) and direct Apoptosis. Additionally, DARZALEX® may play a role in immunomodulation, by depleting CD38-positive regulator immune suppressor cells, and thereby expanding T cells, in patients responding to therapy.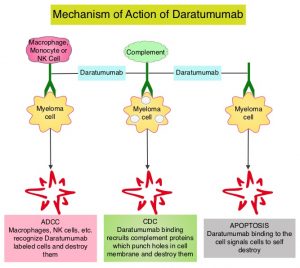
This FDA approval is based on COLUMBA Trial, which is a randomized, open-label, multicenter Phase III study, which included 522 patients with multiple myeloma, who had received at least three prior lines of therapy including a Proteasome Inhibitor (PI) and an immunomodulatory drug (IMiD), or whose disease was refractory to both a PI and an IMiD. Patients were randomly assigned to receive a fixed dose of subcutaneously (SC) administered formulation of DARZALEX® 1800 mg weekly for cycles 1-2, every two weeks for cycles 3-6 and every four weeks for cycle 7 and thereafter (N=263), with the subcutaneous preparation given over 3-5 minutes at alternating left and right abdominal sites. In the intravenous group, patients received DARZALEX® 16 mg/kg IV weekly for cycles 1-2, every two weeks for cycles 3-6 and every four weeks for cycle 7 and thereafter (N=259). Each cycle was 28 days. Treatment in both patient groups was continued until disease progression or unacceptable toxicity. The median age was 67 years and the median number of prior therapies was four in each treatment group. Patient characteristics were similar between the two arms except that more patients in the subcutaneous arm had high-risk cytogenetics (26%) compared with the intravenous group (17%). The median duration of treatment was approximately 5 months, with a median of 6 completed cycles of treatment. The median duration of infusion was consistently 5 minutes at each visit in the subcutaneous group. However, in the IV arm, the first infusion lasted 7 hours, the second infusion was 4.3 hours, and subsequent infusions lasted a median of 3.4 hours. The study co-Primary endpoints were Overall Response Rate (ORR) and pharmacokinetic endpoint of the maximum C-trough on cycle 3, day 1 pre-dose.
At a median follow up of 7.5 months, the ORR was 41% for the subcutaneous administered formulation of DARZALEX® compared to 37% for IV DARZALEX® (P<0.0001). The ORR was similar across all clinically relevant subgroups, including body weight. The ratio of geometric means of C-trough for the SC administered formulation of DARZALEX® over IV DARZALEX® was 108%. The Progression Free Survival was comparable between the SC administered formulation of DARZALEX and the current IV formulation of DARZALEX (HR=0.99; P<0.9258). A lower rate of infusion-related reactions was observed in the group that received the SC DARZALEX® compared to IV DARZALEX® (13% vs. 35%, respectively).
It was concluded that the subcutaneous formulation of DARZALEX® resulted in non-inferior pharmacokinetics and efficacy compared to the current IV formulation, and also importantly offers the potential for a fixed-dose administration, shorter administration times and a lower rate of infusion-related reactions with improved safety profile, in patients with Relapsed or Refractory multiple myeloma.
Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open-label, non-inferiority, randomised, phase 3 trial. Mateos M-V, Nahi H, Legiec W, et al. The Lancet Haematology. Published: March 23, 2020. DOI: https://doi.org/10.1016/S2352-3026(20)30070-3.