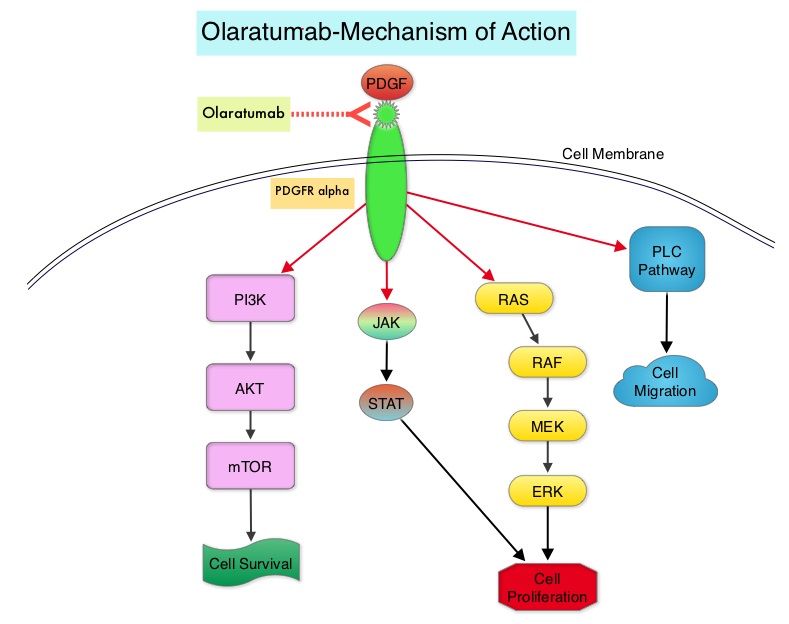
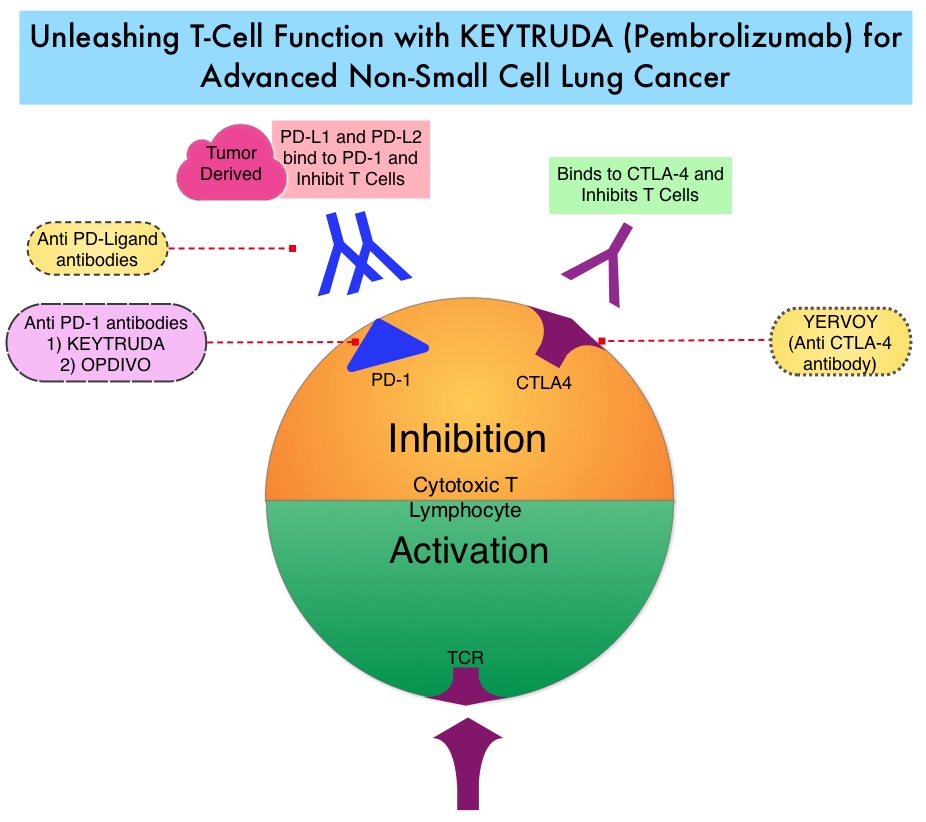
SUMMARY: The FDA on November 10, 2016, approved OPDIVO® (Nivolumab) for the treatment of patients with recurrent or metastatic Squamous Cell Carcinoma of the Head and Neck (SCCHN), with disease progression on or after a Platinum-based therapy. The American Cancer Society estimates that 61,760 people will be diagnosed with Head and Neck cancer in 2016 and 13,190 patients will die of the disease. Patients with recurrent/metastatic Squamous Cell Carcinoma of the Head and Neck have a poor prognosis with a median Overall Survival (OS) of about 13 months with first line therapy and about 6 months or less with later lines of therapy. The treatment paradigm for solid tumors has been rapidly evolving with a better understanding of immune evasion and the role of Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent related to their ability to escape immune surveillance by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies have been developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By blocking the Immune checkpoints, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response.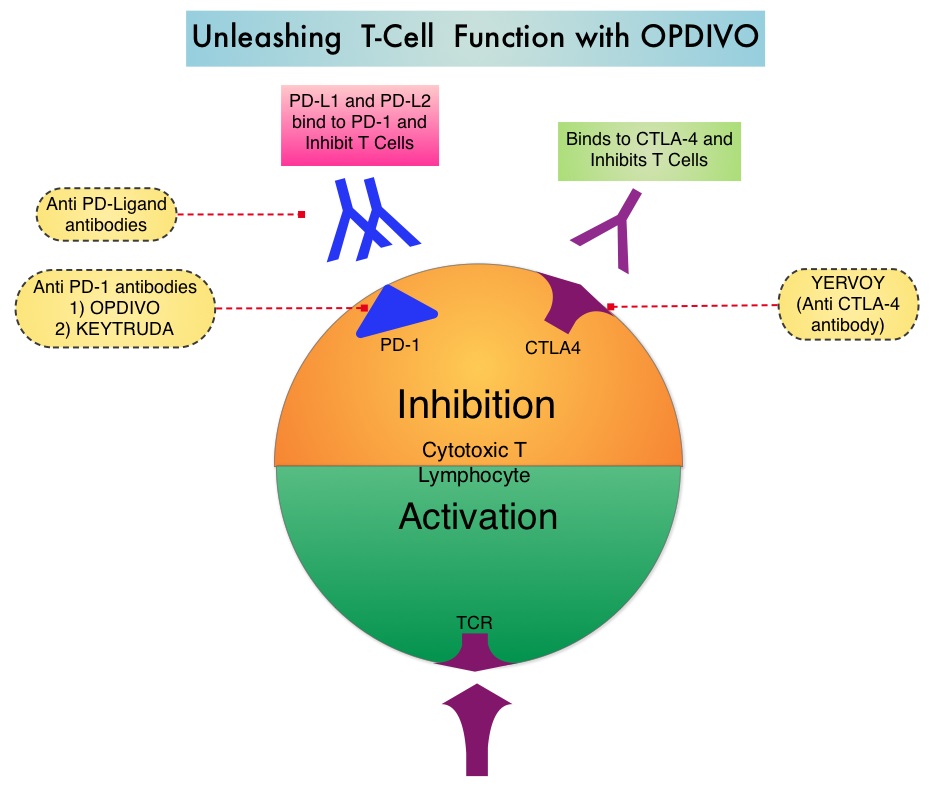
OPDIVO® is an immune checkpoint PD-1 (Programmed cell Death 1) targeted, fully human, immunoglobulin G4 monoclonal antibody that has demonstrated antitumor efficacy in multiple tumor types. The FDA approval of OPDIVO® for the treatment of recurrent or metastatic Squamous Cell Carcinoma of the Head and Neck (SCCHN), was based on the results of CheckMate-141 study which is a randomized, open label, phase III trial. In this study, 361 patients with recurrent Squamous Cell Carcinoma of the Head and Neck (cancer of the oral cavity, pharynx, or larynx), whose disease had progressed within 6 months after Platinum-based chemotherapy, were randomly assigned, in a 2:1 ratio to receive OPDIVO® (N=240) or investigator’s choice of a standard, single agent therapy (N=121). OPDIVO® was administered at a dose of 3 mg/kg every 2 weeks, whereas standard therapy consisted of either weekly Methotrexate at a dose of 40-60 mg/m2 IV, weekly Docetaxel at a dose of 30-40 mg/m2 IV or Cetuximab administered at a loading dose of 400 mg/m2 followed by 250 mg/m2 IV weekly. The median age was 60 years, over 90% had received prior radiation therapy and 54.5% of the patients had received 2 or more lines of prior systemic therapies. The primary end point was Overall Survival and secondary end points included Progression Free Survival, Objective Response Rate, safety, and patient-reported quality of life measures. Prespecified analysis of Overall Survival according to tumor PD-L1 expression and p16 status was also performed.
The median Overall Survival was 7.5 months in the OPDIVO® group versus 5.1 months for the group that received standard therapy and this improvement was statistically significant (HR=0.70; P=0.01). The estimated 1-year survival rate was 36% in the OPDIVO® group and 16.6% with standard therapy. The median Progression Free Survival was 2.0 months with OPDIVO® versus 2.3 months with standard therapy and the rate of Progression Free Survival at 6 months was 19.7% with OPDIVO® versus 9.9% with standard therapy. The Objective Response Rate was 13.3% in the OPDIVO® group versus 5.8% in the standard therapy group. Even though preliminary biomarker analysis suggested that patients with a tumor PD-L1 expression level of 1% or more, or p16-positive tumors, or both, benefited more from OPDIVO® therapy than those whose PD-L1 level was less than 1% or who had p16-negative tumors, these interactions were not statistically significant. Treatment-related grade 3 or 4 adverse events were more common in the standard therapy group (35%) versus OPDIVO® group (13%) and quality of life measures were stable in the OPDIVO® group and were worse for those who received standard therapy.
The authors concluded that OPDIVO® prolonged survival, as compared with standard therapy, among patients with Platinum-refractory Squamous Cell Carcinoma of the Head and Neck and this benefit was accomplished with fewer toxicities, compared with standard therapy. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. Ferris RL, Blumenschein G, Fayette J, et al. N Engl J Med 2016; 375:1856-1867