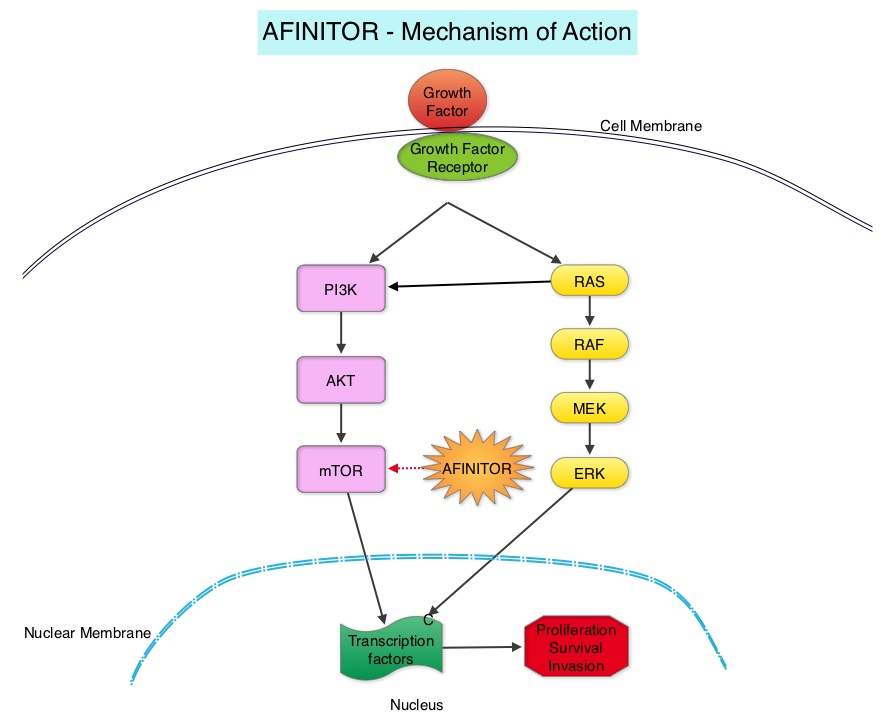
SUMMARY: The American Cancer Society estimates that about 62,700 new cases of kidney cancer will be diagnosed in the United States in 2016 and over 14,000 patients will die from this disease. The VHL (Von Hippel-Lindau) protein is a tumor suppressor gene which is frequently mutated and inactivated in approximately 90% of clear cell Renal Cell Carcinomas (ccRCC). The VHL gene under normal conditions binds to Hypoxia-Inducible Factor (HIF-1 alpha) and facilitates degradation of this factor. Under hypoxic conditions and in patients having biallelic loss of function and mutation of VHL genes, HIF-1alpha is not degraded. Build up of HIF-1 alpha results in increased angiogenesis, increased tumor cell proliferation and survival, as well as metastasis. COMETRIQ® (Cabozantinib) is an oral, small-molecule Tyrosine Kinase Inhibitor (TKI) and inhibits tyrosine kinases including MET, VEGF receptors (VEGFRs), and AXL. Both MET and AXL are up-regulated in Renal Cell Carcinoma as a consequence of VHL inactivation and increased expression of MET and AXL is associated with poor prognosis and development of resistance to VEGFR inhibitors. COMETRIQ® in previous studies has shown objective responses and prolonged disease control in patients with Renal Cell Carcinoma, resistant to VEGFR and mTOR inhibitors. The FDA initially approved COMETRIQ® in 2012, for treatment of patients with metastatic Medullary Thyroid Cancer. AFINITOR® (Everolimus) is a specific inhibitor of mTOR (Mammalian Target of Rapamycin), which is a serine/threonine kinase, and is a standard treatment for patients who progress on a VEGFR-targeted therapy.
The METEOR is a phase III trial in which 658 patients were randomized 1:1 to receive COMETRIQ® 60 mg PO daily or AFINITOR® 10 mg PO daily. Treatment was continued until disease progression or unacceptable toxicities. Enrolled patients had advanced clear cell Renal Cell Carcinoma and were stratified by MSKCC (Memorial Sloan Kettering Cancer Center) prognostic criteria and number of prior therapies with VEGFR TKIs. Of the enrolled patients in the COMETRIQ® group, 43% of the patients were considered favorable, 43% intermediate and 14% poor risk, by MSKCC criteria. Seventy three percent (73%) of the patients had one prior therapy with VEGFR TKIs and 27% of the patients had 2 or more prior therapies with VEGFR TKIs. To be eligible, patients must have progressed during treatment or within 6 months of the last dose of their most recent VEGFR TKI. Prior therapies included cytokines, chemotherapy, and monoclonal antibodies, including those targeting VEGF, the Programmed Death 1 (PD-1) receptor or its ligand PD-L1. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Overall Survival (OS) and Objective Response Rate (ORR). At the time of preplanned interim analysis which included the first 375 patient who underwent randomization, the primary end point of PFS was met, with a significant improvement in PFS with COMETRIQ® compared to AFINITOR® (7.4 months vs 3.8 months; HR=0.58; P< 0.001). In addition, there was a significant improvement in ORR with COMETRIQ® (21% vs 5%; P<0.001) and a trend for improved OS. (N Engl J Med 2015; 373:1814-1823).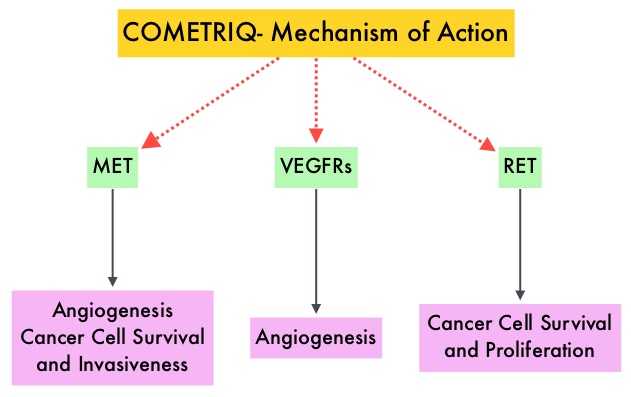
In this updated analysis, the authors provided a detailed analysis of the clinical activity of COMETRIQ compared to AFINITOR® across the various patient subgroups. For all 658 patients enrolled, the PFS data was comparable to the interim analysis data, favoring COMETRIQ® (7.4 months versus 3.9 months (HR=0.52; P<0.001). When subgroup analysis was performed in patients with 3 or more metastases sites, the median PFS was 7.3 months versus 3.7 months for COMETRIQ® vs AFINITOR® respectively (HR=0.38). The risk of disease progression was reduced with COMETRIQ® by 74% in patients with visceral and bone metastases compared to AFINITOR® (5.6 vs 1.9 months; HR=0.26). The PFS benefit with COMETRIQ® was not impacted by prior therapy with VEGFR TKIs. However, patients who received prior VEGFR TKI therapy with SUTENT® (Sunitinib) benefited the most with COMETRIQ® (median PFS 9.1 vs 3.7 months (HR=0.43), whereas the PFS benefit with COMETRIQ® for those who received prior VOTRIENT® (Pazopanib) was 7.4 vs 5.1 months (HR=0.67). The PFS benefit was also significantly better in the COMETRIQ® group, for patients previously treated with an anti–PD-1/PD-L1 agents, compared to AFINITOR® (HR=0.22). In the MSKCC poor risk group, the benefit with COMETRIQ® was 5.4 versus 3.5 months with AFINITOR® (HR=0.70). The most common serious toxicities AEs in the COMETRIQ® group were abdominal pain, pleural effusion and diarrhea, whereas in the AFINITOR® arm, the most common serious toxicities were anemia, dyspnea and pneumonia.
The authors concluded that COMETRIQ® is associated with longer Progression Free Survival compared with AFINITOR®, in patients with Renal Cell Carcinoma, following progression on prior VEGFR inhibitor therapy. COMETRIQ® may help overcome treatment resistance and benefits all subgroups of patients with advanced Renal Cell Carcinoma. Studies are underway combining COMETRIQ® with Immune checkpoint inhibitors. Subgroup analyses of METEOR, a randomized phase 3 trial of cabozantinib versus everolimus in patients (pts) with advanced renal cell carcinoma (RCC). Escudier BJ, Motzer RJ, Powles T, et al. J Clin Oncol 34, 2016 (suppl 2S; abstr 499)