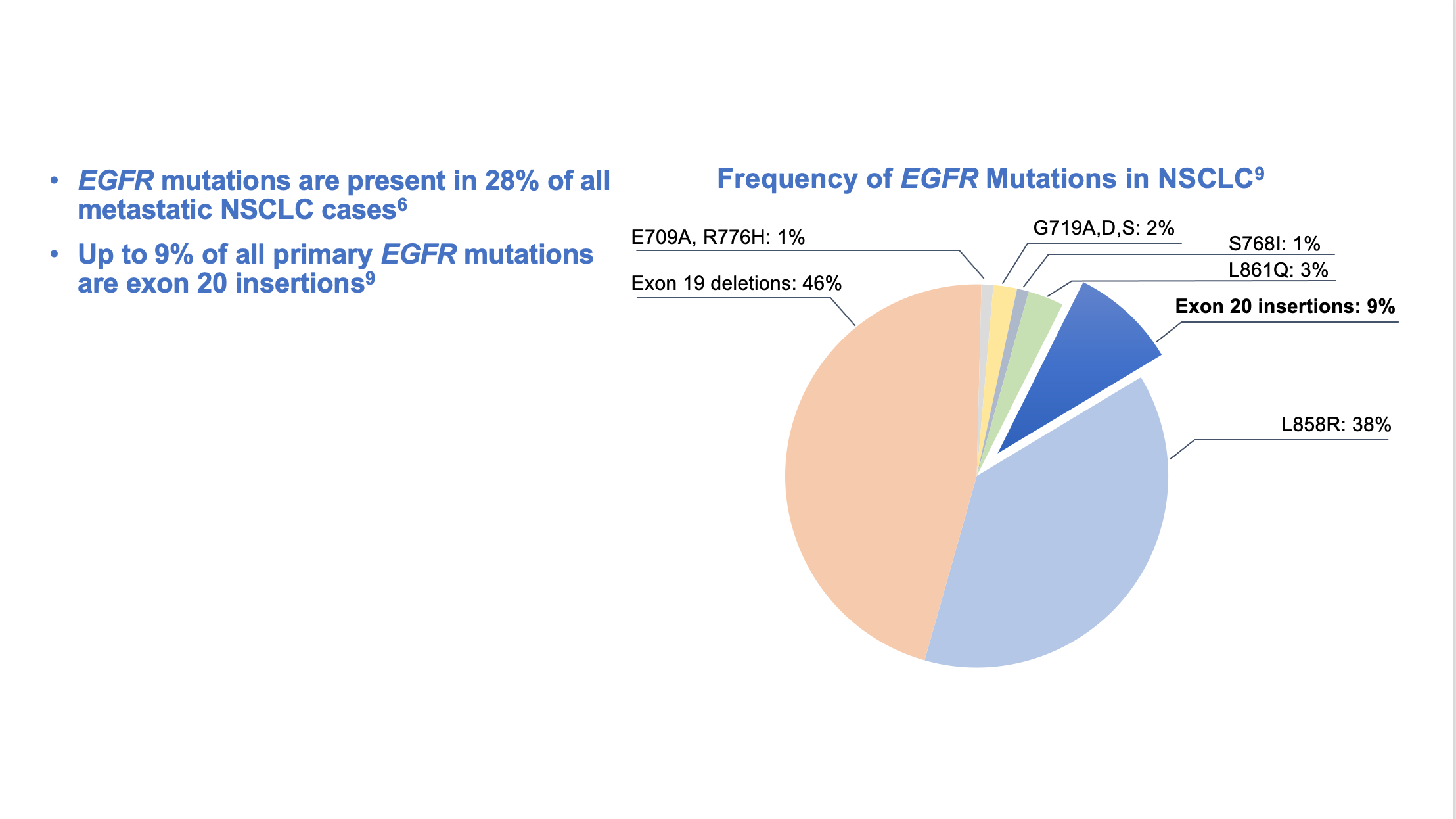
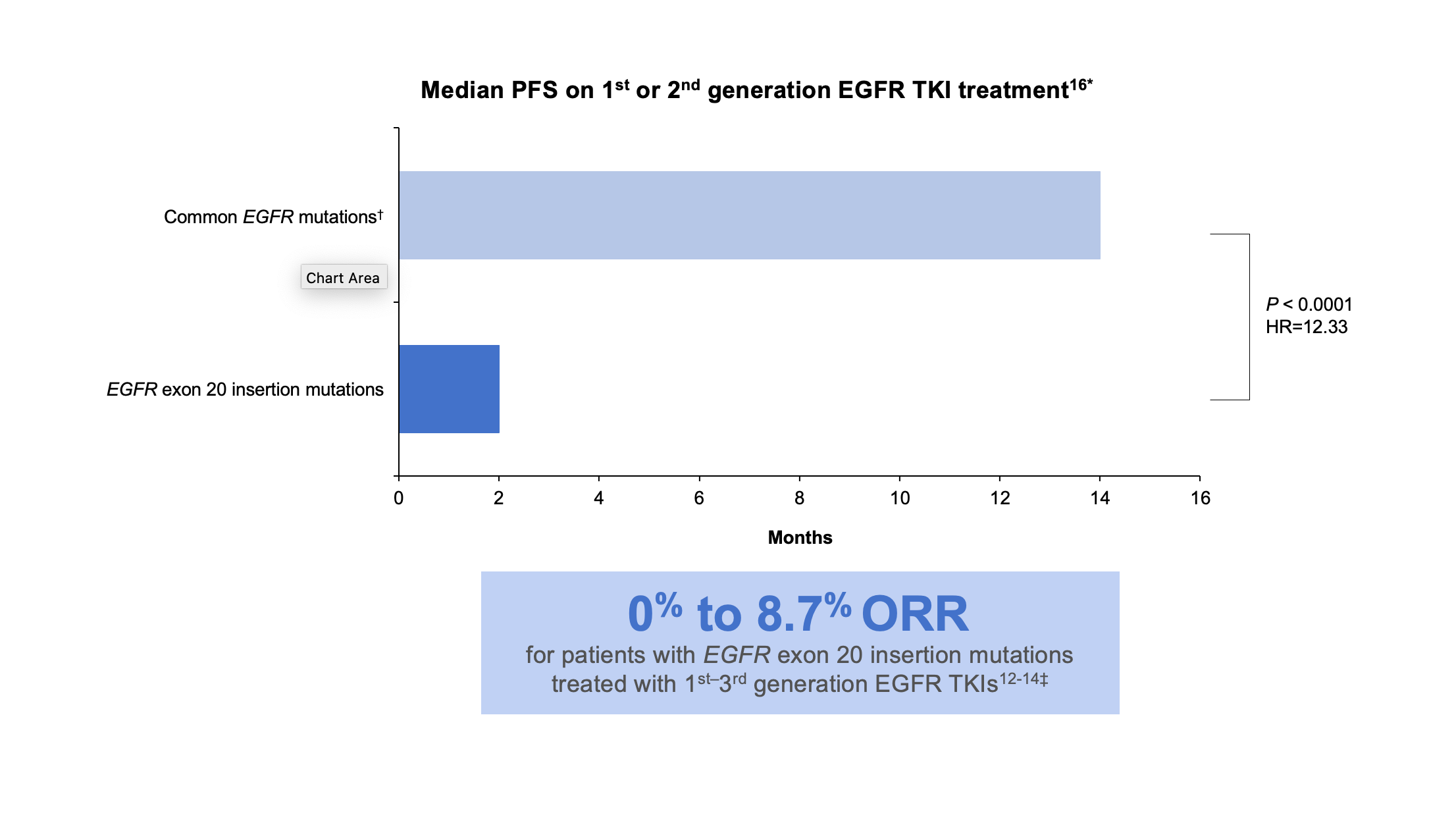
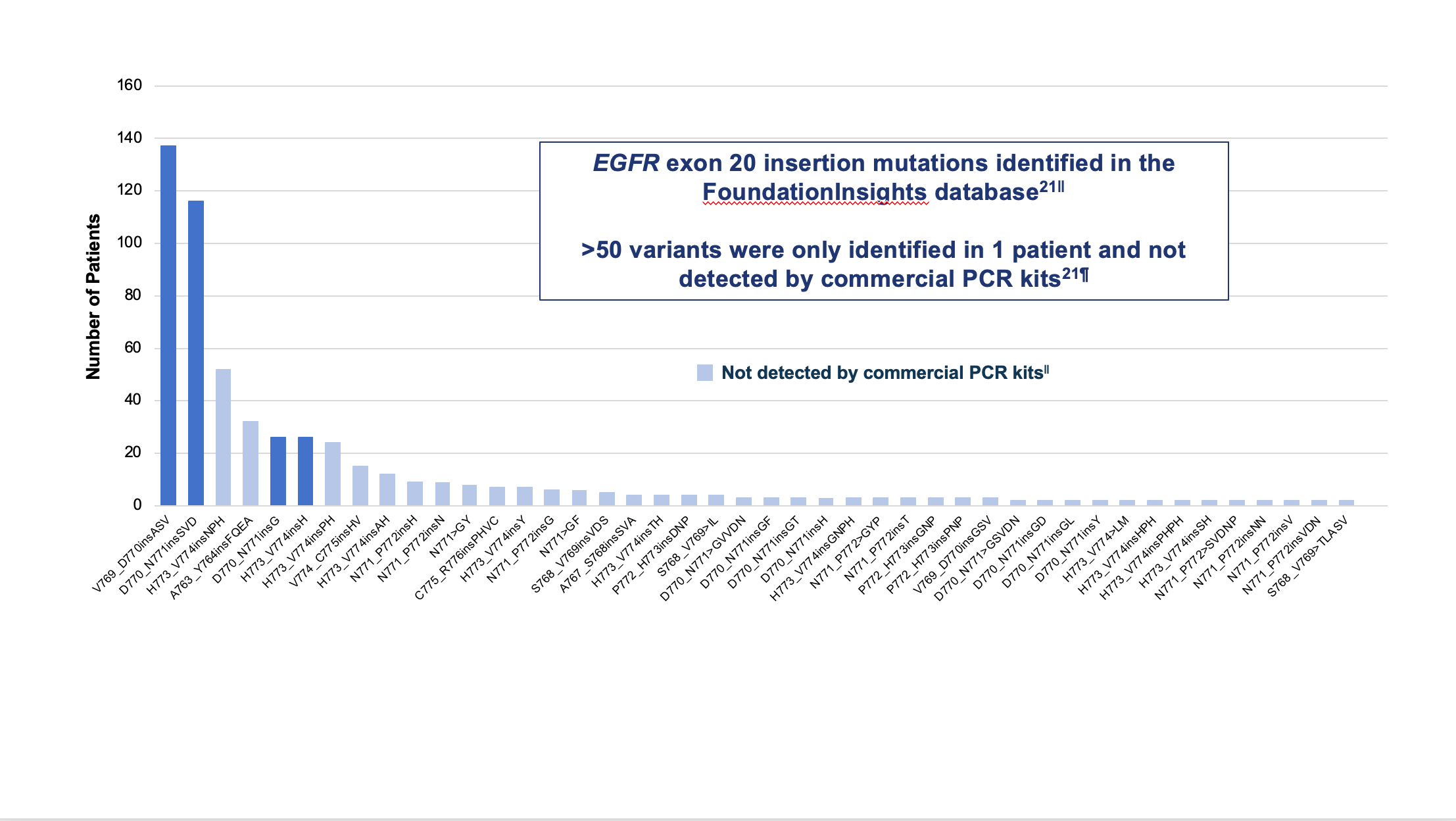
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 21% of all cancer deaths. The American Cancer Society estimates that for 2023, about 238,340 new cases of lung cancer will be diagnosed and 127,070 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Low Dose CT (LDCT) screening for lung cancer resulted in a 20% reduction in mortality In the National Lung Screening Trial (NLST). The USPSTF expanded the criteria for lung cancer screening in 2021 and recommended annual screening with Low-Dose CT for adults aged 50-80 years who have a 20 pack-year smoking history and currently smoke or have quit within the past 15 years. Approximately 15% of patients present with early stage (T1-2 N0) disease, and these numbers are likely to increase with the more rigorous implementation of lung cancer screening programs.
Surgical resection is the primary treatment for approximately 30% of patients with NSCLC who present with early Stage (I–IIIA) disease. Pneumonectomy is rarely performed due to unacceptably high mortality rate. Lobectomy has been the standard of surgical care for patients with clinical T1N0 NSCLC since the mid 1990s. This was based on the results of a randomized trial comparing Lobectomy with Sublobar resection in patients with clinical T1N0 NSCLC. In this trial, the frequency of local recurrence was three times higher with Sublobar resection compared with Lobectomy, and lung cancer-related mortality was 50% higher with Sublobar resection.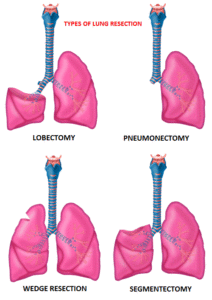
Sublobar resection includes Wedge resection and Segmentectomy. In Wedge resection, the lung tumor is removed with a surrounding margin of normal lung tissue, and is not an anatomical resection. Segmentectomy, unlike Wedge resection, is an anatomical resection that usually includes one or more pulmonary parenchymal segments with the dissection of intraparenchymal and hilar lymph nodes. Advances in imaging as well as staging by means of mediastinoscopy, and routine intraoperative lymphadenectomy has enabled the identification of small, peripheral NSCLCs for which Sublobar resection was potentially appropriate. Sublobar resection was considered a “compromise operation” in selected high risk patients with early stage lung cancer. With the approval of lung cancer screening in high risk individuals and subsequent detection of small tumors, Sublobar resections have been on the rise and may be the preferred surgical option, even in good-risk patients, in many institutions. Sublobar resection preserves pulmonary function and leaves open more treatment options for NSCLC patients, who remain at high risk for metachronous primary NSCLC, following curative intervention for their first NSCLC.
With the implementation of CT-based lung cancer screening recently, lung cancers are likely detected at a very early stage (T1a-bN0; 2 cm or less, node negative tumors). Further, Adenocarcinoma now is the most frequent histologic subtype of lung cancer and present as peripherally located tumors. Advances in preoperative staging such as endobronchial ultrasonography, have improved patient selection for treatment. Majority of surgical resections are now performed by means of video or robotic-assisted thoracic surgery. This has improved postoperative outcomes, with significant reduction in perioperative morbidity, mortality and median length of hospital stay after either Sublobar resection or Lobectomy.
The authors in this study reported the results of a randomized international trial comparing Sublobar resection (wedge resection or segmentectomy) with Lobectomy, in patients with clinical Stage IA NSCLC, with a tumor size of 2 cm or less. Cancer and Leukemia Group B (CALGB) 140503 was a multicenter, international, randomized, noninferiority, Phase III trial, involving patients with NSCLC clinically staged as T1aN0. In this study, a total of 697 patients, after intraoperative confirmation of node-negative disease, were randomly assigned to undergo either Sublobar resection (N=340) or Lobar resection (N=357). Of the 340 patients assigned to Sublobar resection, 201 (59.1%) underwent wedge resection and 129 (37.9%) underwent an anatomical segmental resection. Wedge resection was allowed in the current trial as it is the most frequently practiced method of Sublobar resection in North America and Europe and its inclusion would make the trial more representative of a “real world” setting. The median patient age was 68 years. Approximately 50% of patients had tumor size 1.0-1.5 cm, 40% had tumor size 1.5-2.0 cm, and two thirds of the patients had Adenocarcinoma histology. Over 90% of the patients were current or former smokers. The Primary end point was Disease-Free Survival (DFS), defined as the time between randomization and disease recurrence or death from any cause. Secondary end points included Overall Survival (OS), locoregional and systemic recurrence, and pulmonary functions.
After a median follow up of 7 years, Sublobar resection was noninferior to Lobar resection for DFS (HR for disease recurrence or death=1.01). The 5-year DFS was 63.6% after Sublobar resection and 64.1% after Lobar resection. The Overall Survival after Sublobar resection was similar to that after Lobar resection (HR for death, 0.95). The 5-year OS was 80.3% after Sublobar resection and 78.9% after Lobar resection. No substantial difference was seen between the two groups in the incidence of locoregional or distant recurrence. At 6 months postoperatively, pulmonary functions favored the Sublobar resection group.
It was concluded that Sublobar resection by either anatomical segmentectomy or wedge resection, for patients with peripheral NSCLC with a tumor size of 2 cm or less and pathologically confirmed node-negative disease in the hilar and mediastinal lymph nodes, was non inferior to Lobectomy, with respect to Disease Free Survival and with similar Overall Survival, and is an effective management approach for this subgroup of patients with NSCLC.
Lobar or Sublobar Resection for Peripheral Stage IA Non–Small-Cell Lung Cancer. Altorki N, Wang X, Kozono D, et al. N Engl J Med 2023; 388:489-498