SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
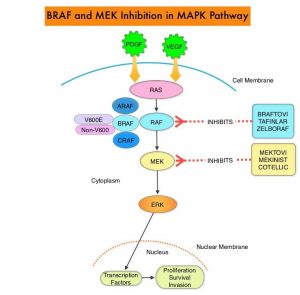
In addition to the well characterized gene fusions involving ALK and ROS1 in NSCLC, genetic alterations involving other kinases including EGFR, BRAF, RET, NTRK, MET, HER2 are all additional established targetable drivers. These genetic alterations are generally mutually exclusive, with no more than one predominant driver in any given cancer. The hallmark of all of these genetic alterations is oncogene addiction, in which cancers are driven primarily, or even exclusively, by aberrant oncogene signaling, and are highly susceptible to small molecule inhibitors. Patients with nonsquamous NSCLC should therefore be tested for Actionable Driver Oncogenes, as highly effective treatments may be available for these patients. Nonetheless, Single Gene Testing for EGFR and ALK is more common in the US rather than broad multigene panel testing with Next-Generation Sequencing.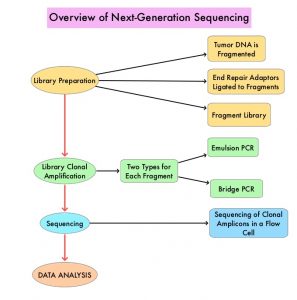
Next-Generation Sequencing (NGS) platforms or second-generation sequencing, unlike the first-generation sequencing, known as Sanger sequencing, perform massively parallel sequencing, which allows sequencing of millions of fragments of DNA from a single sample. With this high-throughput sequencing, the entire genome can be sequenced in less than 24 hours. There are a number of different NGS platforms using different sequencing technologies and NGS can be used to sequence and systematically study the cancer genomes in their entirety or specific areas of interest in the genome or small numbers of individual genes. Recently reported genomic profiling studies, performed in patients with advanced cancer suggest that actionable mutations are found in 20-40% of patients’ tumors.
The authors in this study used a decision analytic model they had developed, and compared the value of broad NGS-based testing, to Single Gene Testing (SGT), in patients with nonsquamous NSCLC, and discussed their implications for the US population. The authors noted that Single Gene Testing for EGFR and ALK is relatively common (>80%) in the US, whereas testing for less common Actionable Driver Oncogenes is rare. The broader NGS Actionable Driver Oncogene panel includes EGFR, ALK, ROS1, BRAF, RET, MET, NTRK. The authors took into consideration reimbursement by CMS for broad NGS-based testing ($627.50), reimbursement for Single Gene testing (EGFR+ALK $732.30), and the cost of treatment for 2 years at $10K/year ($20,000). The expected prevalence of Actionable Driver Oncogenes among non squamous NSCLC patients, as well as survival outcomes of patients, in the presence versus absence of an Actionable Driver Oncogenes treatment strategy, was calculated based on current literature. The number of eligible patients with nonsquamous NSCLC, for testing in the US, were 89,000 (N=89,000). The estimated number of patients with Actionable Driver Oncogenes (EGFR, ALK, ROS1, BRAF, RET, MET, NTRK) was 26,300 (N=26,300). The goal of this study was to measure the cost and value differences when one chose to run a Single Gene Testing (narrow genomics panel), which included interrogation for either EGFR or ALK, versus a broader NGS panel. The potential value of each testing approach was measured based on Life Years Gained (LYG) and the cost per LYG. (Life Years gained is a modified mortality measure where remaining life expectancy is taken into account).
It was noted that a broad NGS approach to test for genetic alterations resulted in additional Life Year Gains with cost savings, compared to Single Gene Testing for EGFR or ALK. This analytical model suggested that at the current 80% testing rate, replacing Single Gene Testing with NGS would result in an additional 21,019 Life Year Gained, with reduced cost per LYG of $599. Increasing testing from 80% to 100% of eligible patients would further increase the Life Year Gained by 15,017. If 100% of eligible patients were tested with NGS and every identified patient received treatment, the cost per Life Year Gained with this strategy would be $16,641.57.
According to this decision model, the estimated median survival and 5-year survival for a patient who was tested with NGS, followed by a highly effective therapy selected on the basis of that alteration, would be 39 months and 25%, respectively. For a patient who had an Actionable Driver Oncogene that was not identified by Single Gene Testing, the estimated median survival would be 14 months and 5-year survival would be 5%. This analysis suggested that not running broad multigene NGS panel routinely for eligible patients, and only using Single Gene Testing could be a missed opportunity, as actionable mutations would be missed and patients may not get the most effective therapy for their disease.
The authors concluded that based on their decision analytic model, when highly effective therapy is available to all identified patients with Actionable Driver Oncogenes, broad NGS testing, compared to Single Gene Testing for EGFR or ALK, leads to large gains in Life Years, at reduced cost per Life Year Gained, compared to Single Gene Testing. This model supports universal NGS testing of all patients with advanced nonsquamous NSCLC.
A model comparing the value of broad next-gen sequencing (NGS)-based testing to single gene testing (SGT) in patients with nonsquamous non-small cell lung cancer (NSCLC) in the United States. Pennell NA, Zhou J, Hobbs B. J Clin Oncol 38: 2020 (suppl; abstr 9529)