SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2019 about 228,150 new cases of lung cancer will be diagnosed and 142,670 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas.
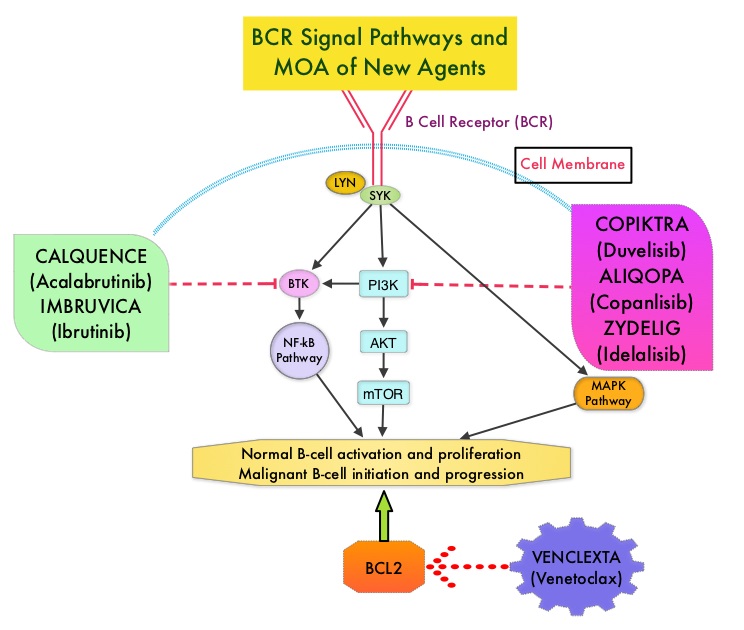
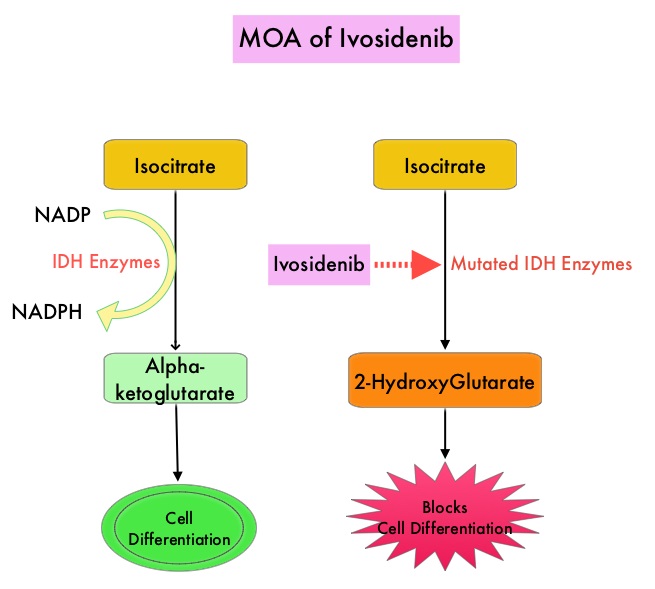
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression, and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®.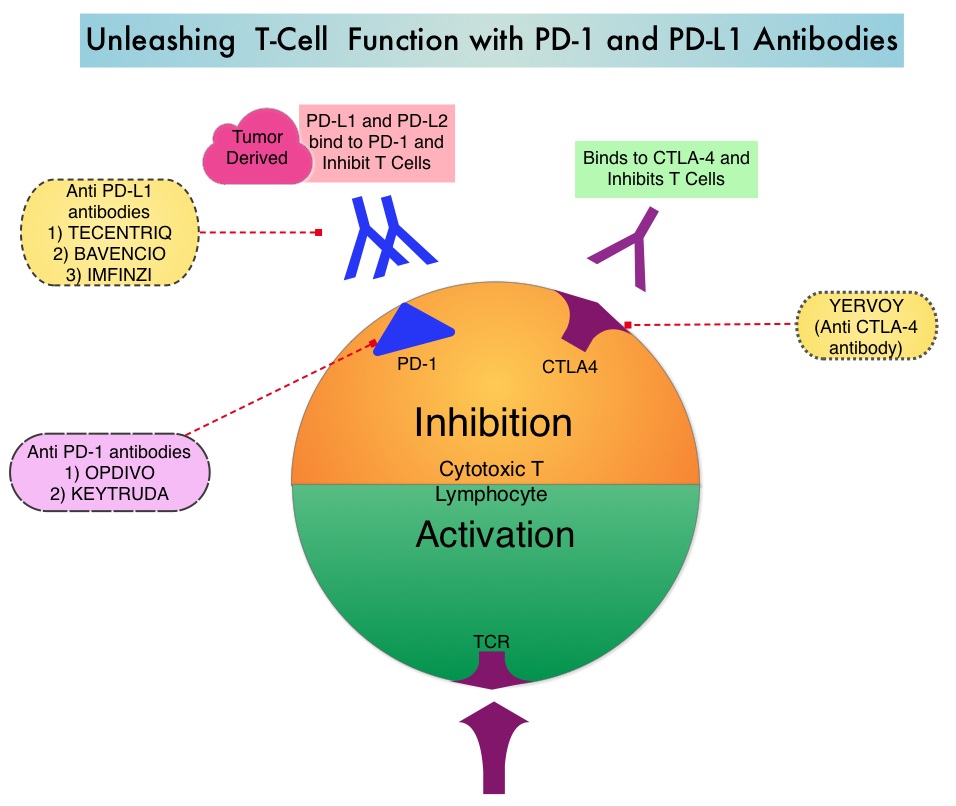
The FDA approved KEYTRUDA® for the first-line treatment of patients with Stage III Non-Small Cell Lung Cancer (NSCLC) who are not candidates for surgical resection or definitive chemoradiation, as well as those with metastatic NSCLC whose tumors express PD-L1 (Tumor Proportion Score-TPS of 1% or more), as determined by an FDA-approved test. KEYTRUDA® is also approved for the first-line treatment of advanced NSCLC with high PD-L1 expression (Tumor Proportion Score of 50% or more), based on KEYNOTE-024 trial, as well as in combination with Pemetrexed and Carboplatin, as first-line treatment of patients with metastatic non-squamous NSCLC, based on KEYNOTE-021 study. It is also indicated for previously treated advanced NSCLC with a much lower level of PD-L1 expression such as PD-L1 Tumor Proportion Score of 1% or higher, based on KEYNOTE-010 trial.
The authors in this publication presented the 5-year Overall Survival (OS) for patients enrolled in the Phase 1b KEYNOTE-001 study, which was the first trial evaluating KEYTRUDA® in advanced NSCLC. In this trial, 550 patients were enrolled of whom 101 patients were treatment-naïve (N=101) and 449 patients were previously treated (N=449). Patients received KEYTRUDA® 2 mg/kg IV every 3 weeks or KEYTRUDA® 10 mg/kg IV every 2 or 3 weeks. The protocol in the recent years was changed to a straight dose of KEYTRUDA® 200 mg IV every 3 weeks, which is the typical regimen used in clinical practice. The Primary endpoint was Objective Response Rate (ORR). Secondary endpoints included Progression Free Survival (PFS), Overall Survival (OS) and Duration of Response (DOR). The median follow up was 60.6 months and 18% of participants (N=100) were still alive at that point.
The 5-year OS in the treatment-naïve patients (N=101) was 23.2% and 15.5% in previously treated patients (N=449). In treatment-naive patients, the 5-year OS rate among patients whose tumors expressed PD-L1 expression of 50% or more was 29.6%, compared with 15.7% with PD-L1 expression levels below 50%. In patients who had received previous treatment, the 5-year OS rate among patients whose tumors expressed PD-L1 expression of 50% or more was 25% compared with 12.6% with PD-L1 expression levels between 1% and 49%. Only 3.5% of people with PD-L1 expression levels below 1% were alive after 5 years. The investigator-reported ORR was 41.6% in treatment-naïve patients and 22.9% in previously treated patients. Median Duration of Response was 16.8 months and 38.9 months respectively. Immune-mediated adverse events were reported in 17% of patients at 5 years. Hypothyroidism was the most commonly reported immune-mediated adverse event, followed by pneumonitis, hyperthyroidism and skin toxicities.
It was concluded that the 5-year data from the KEYNOTE-001 trial showed that treatment with KEYTRUDA® was safe and effective and substantially increased Overall Survival in patients with advanced NSCLC. These data provide the longest efficacy and safety follow-up for NSCLC patients treated with KEYTRUDA®. Five-year long-term overall survival for patients with advanced NSCLC treated with pembrolizumab: Results from KEYNOTE-001. Garon EB, Hellmann MD, Costa EC, et al. J Clin Oncol. 2019;37(suppl; abstract LBA9015).
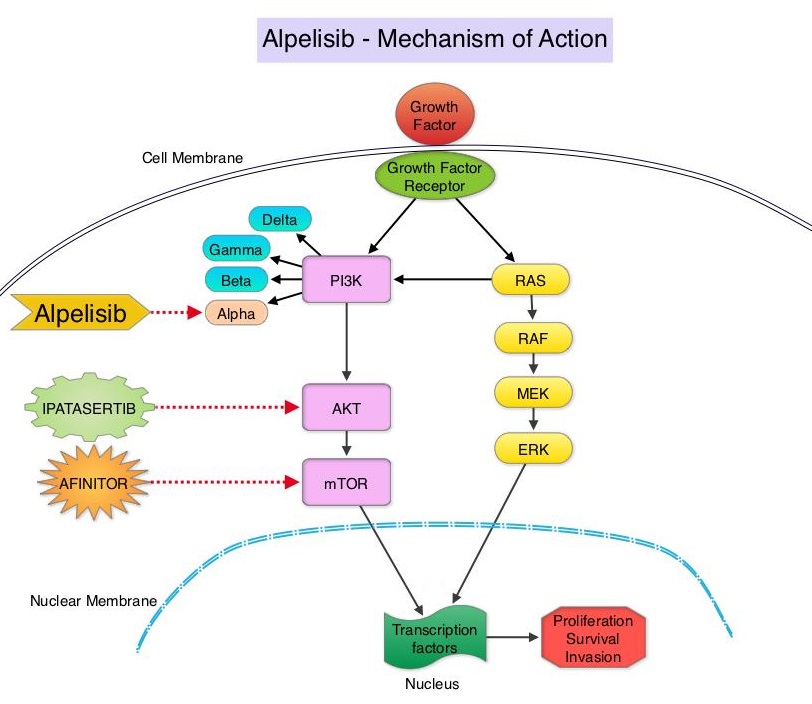
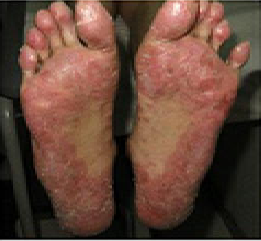
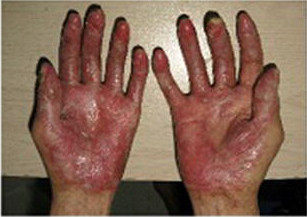 In previously published studies, JAKAFI® when used in patients with refractory GVHD demonstrated an Overall Response Rate of 85% in acute or chronic GVHD, with a 25% Complete Remission rate.
In previously published studies, JAKAFI® when used in patients with refractory GVHD demonstrated an Overall Response Rate of 85% in acute or chronic GVHD, with a 25% Complete Remission rate.