SUMMARY: The FDA on April 30, 2018, granted regular approval to TAFINLAR® (Dabrafenib) and MEKINIST® (Trametinib), in combination, for the adjuvant treatment of patients with melanoma with BRAF V600E or V600K mutations, as detected by an FDA-approved test, and involvement of lymph node(s), following complete resection. It is estimated that in the US, approximately 91,270 new cases of melanoma will be diagnosed in 2018 and about 9,320 patients are expected to die of the disease. The incidence of melanoma has been on the rise for the past three decades. Surgical resection with a curative intent is the standard of care for patients with early stage melanoma, with a 5-year survival rate of 98% for stage I disease and 90% for stage II disease. Stage III malignant melanoma however is a heterogeneous disease, and the risk of recurrence is dependent on the number of positive nodes, as well as presence of palpable versus microscopic nodal disease. Further, patients with a metastatic focus of more than 1 mm in greatest dimension in the affected lymph node, have a significantly higher risk of recurrence or death than those with a metastasis of 1 mm or less. Patients with stage IIIA disease have a disease-specific survival rate of 78% whereas those patients with stage IIIB and stage IIIC disease have disease-specific survival rates of 59% and 40% respectively.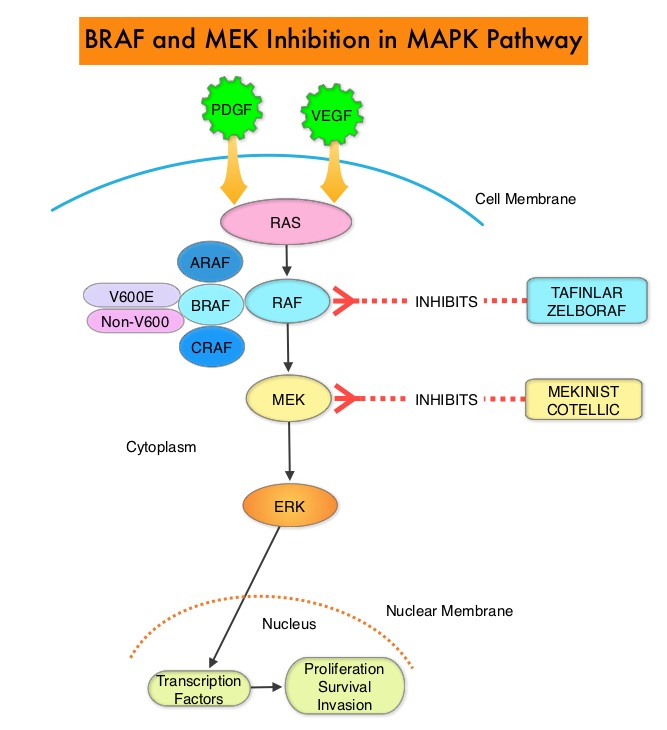
The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role, regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6-8% of all malignancies. The most common BRAF mutation in melanoma is at the V600E/K site and is detected in approximately 50% of melanomas and result in constitutive activation of the MAPK pathway.
TAFINLAR®,is a selective oral BRAF inhibitor and MEKINIST® is a potent and selective inhibitor of MEK gene, which is downstream from RAF in the MAPK pathway. In patients with BRAF V600 mutation-positive unresectable or metastatic melanoma, a combination of TAFINLAR® and MEKINIST® resulted in a median Overall Survival (OS) of more than 2 years, with approximately 20% of the patients remaining progression free at 3 years. These encouraging results led to the study of this combination in patients with stage III melanoma, with BRAFV600E or V600K mutations, after complete surgical resection.
This FDA approval was based on COMBI-AD, an international, multi-center, randomized, double-blind, placebo-controlled, phase III trial, in which 870 patients with completely resected, stage III melanoma and with BRAF V600E or V600K mutations were enrolled. Patients were randomly assigned in a 1:1 to receive TAFINLAR® 150 mg orally twice daily in combination with MEKINIST® 2 mg orally once daily (N=438) or two matched placebos (N=432). Treatment was given for 12 months. Eligible patients had undergone completion lymphadenectomy, with no clinical or radiographic evidence of residual regional node disease. None of the patients had received previous systemic anticancer treatment or radiotherapy for melanoma. BRAF V600 mutation status was confirmed in primary tumor tissue or lymph node tissue by a central reference laboratory. The median age was 50 years. Both treatment groups were well balanced and 18% had stage IIIA disease, 41% had stage IIIB disease, and 40% had stage IIIC disease. Of the enrolled patients, 91% had a BRAF V600E mutation, and 9% had a BRAF V600K mutation. The Primary end point was Relapse Free Survival (RFS) and Secondary end points included Overall Survival (OS), Distant metastasis-free survival, Freedom from relapse, and Safety.
At a median follow up of 2.8 years, the estimated 3-year RFS rate was 58% in the combination therapy group and 39% in the placebo group (HR=0.47; P<0.001), and this represented a 53% lower risk of relapse. At the time of this analysis, median RFS rate had not yet been reached in the combination therapy group and was 16.6 months in the placebo group. The improved RFS benefit with the combination therapy was consistent across patient subgroups, regardless of lymph node involvement or primary tumor ulceration. The risk of distant metastases or death was reduced by 49% with the combination therapy versus placebo (HR=0.51; P<0.001). The safety profile of TAFINLAR® plus MEKINIST® was consistent with that observed with the combination, in patients with metastatic melanoma, and the common side effects were pyrexia, fatigue, nausea, vomiting, diarrhea, headache, rash, arthralgia, and myalgia.
It was concluded that adjuvant combination therapy with TAFINLAR® plus MEKINIST® in patients with stage III melanoma with BRAF V600E or V600K mutations, resulted in a significantly lower risk of recurrence, compared to placebo, with no new adverse events. With more than half the patients with stage III melanoma having a recurrence after surgery, this first effective oral targeted combination therapy, will be an important adjuvant treatment option. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. Long GV, Hauschild A, Santinami M, et al. N Engl J Med 2017; 377:1813-1823
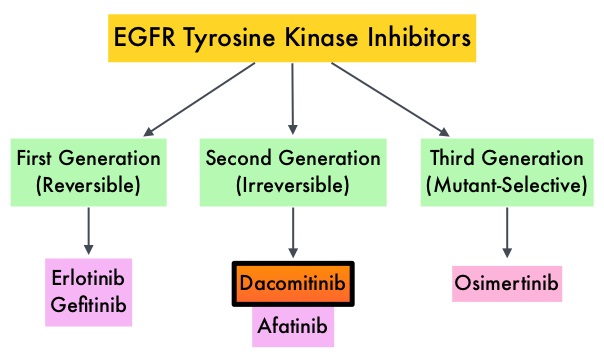 EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. T
EGFR-Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60-70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations. However, majority of these patients experience disease progression within 9-14 months. T