SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and about 40,920 women will die of the disease. Circulating Tumor Cells (CTCs) are epithelial cells that are shed into the circulation from a primary or metastatic tumor. After being shed, CTCs can remain in the circulation or undergo apoptosis. Evaluation of CTCs during the course of disease has prognostic value. Because of the very low concentrations of CTCs (1 CTC in the background of millions of normal hematopoietic cells) in the peripheral blood, different technologies have been developed that will allow enrichment and detection of these CTCs. One such technology is the CELLSEARCH® system which is the first FDA-approved test for CTC assessment, in the peripheral blood of patients with breast cancer. This automated system is able to enrich the peripheral blood sample with CTCs and the cells then are fluorescently stained for CytoKeratins (CK8,18 and 19), Common Leukocyte Antigen (CD45) and a nuclear dye (DAPI). CTCs are identified when they are CK positive, CD45 negative and DAPI positive. In essence, CTC assessment is a real time, peripheral blood evaluation (“Liquid Biopsy”) in breast cancer patients.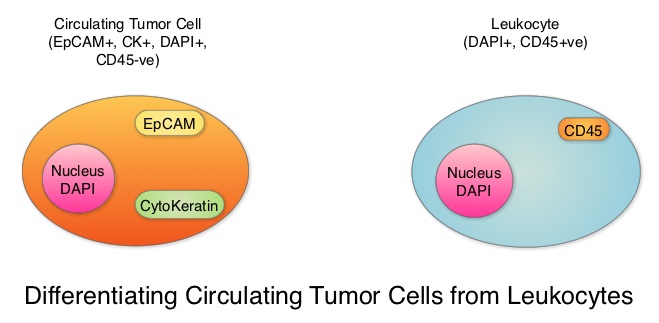
In women with Hormone Receptor (HR)-positive breast cancer, late recurrences occurring 5 or more years after diagnosis, account for approximately 50% of recurrences. In a meta-analysis from the EBCTCG (Early Breast Cancer Trialists’ Collaborative Group) group, the 10-year risk of recurrence after 5 years of endocrine therapy was found to be 5% in patients with node negative breast cancer, 10% in patients with 1-3 positive lymph nodes, and 22% in patients with 4-9 positive lymph nodes. Late recurrence in patients with HR-positive breast cancer has therefore become a major concern, as this risk for recurrence and death from breast cancer can be ongoing for at least 20 years after the original diagnosis..
The blood and tumor samples of patients are valuable for research and allow the exploration of possible tumor and host-related factors contributing to recurrence. The Coalition of Cancer Cooperative Groups, a non-profit member organization of the National Cancer Institute-sponsored Cooperative Groups, in 2013, created the North American Breast Cancer Groups (NABCG) Biospecimen Bank for Determinants of Late Relapse in Operable Breast Cancer. The purpose for the collection and analysis of samples is, to improve our understanding of breast cancer, and identify which patients are most likely to benefit from specific therapies. The NABCG Biospecimen Bank remains a unique resource of archived primary and metastatic tumor tissue, blood, and DNA, collected from more than 15,000 women, who participated in two large cooperative group cancer treatment trials, TAILORx and E5103.
The authors in this analysis included 546 patients with HER-2-negative, stage II to stage III breast cancer from the E5103 clinical trial, with no clinical evidence of recurrence between 4.5 to 7.5 years following their initial diagnosis. ECOG-ACRIN clinical trial E5103 is a randomized phase III trial, which evaluated the addition of Bevacizumab (AVASTIN®), a Vascular Endothelial Growth Factor (VEGF) inhibitor, to adjuvant chemotherapy following surgery, in patients with lymph node-positive or high-risk, lymph node-negative breast cancer. All patients had undergone surgery and adjuvant chemotherapy as well as endocrine therapy for at least 5 years, if hormone receptor positive. The CTCs from the blood samples of these patients were measured using the CELLSEARCH® CTC assay. None of the patients had clinical evidence of recurrence at the time of this analysis. More than half of the patients (56%) were 50 years or older, 59% of the patients had a tumor 2 cm or larger, 73% had positive lymph nodes and 65% had Hormone Receptor (HR) positive disease. This Primary endpoint of this study was time to recurrence and the primary focus in this analysis was on the group with Hormone Receptor positive disease.
After a median follow up of 1.6 years, 4.8% of patients had a positive CTC assay result overall (1 or more cells/7.5 mL in the blood). Among those with Hormone Receptor positive breast cancer, 5.1% had a positive CTC assay result whereas positive CTC assay result was noted in 4.1% of patients with hormone receptor negative disease. Of the 353 patients with HR-positive disease in this analysis, 4% had a recurrence whereas only 0.5% with HR-negative disease had a local recurrence. The Hazard Ratio for patients with CTC negative results compared with CTC positive results was 21.7 (P<0.001). A positive CTC assay result was associated with a nearly 21.7 fold increased risk of breast cancer recurrence in patients with HR-positive disease. A positive CTC assay was not associated with recurrence in the HR-negative group. The Positive Predictive Value of a positive CTC assay for recurrence at 2 years, in patients with HR-positive disease was 35%, and the Negative Predictive Value for patients in this cohort was 98%.
It was concluded that detection of positive Circulating Tumor Cells in the blood, 5 years after breast cancer diagnosis, was associated with an increased risk for late recurrence, in women with HR-positive, HER2-negative breast cancer. Sparano JA, O’Neill A, Alpaugh K, et al. Circulating tumor cells and late recurrence of breast cancer. Presented at: 2017 San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, TX. Abstract GS6-03.