SUMMARY: The American Cancer Society estimates that in 2025, about 80,350 people will be diagnosed with Non Hodgkin Lymphoma (NHL) in the United States and about 19,390 individuals will die of this disease. Indolent Non-Hodgkin Lymphomas are mature B cell lymphoproliferative disorders and include Follicular Lymphoma, Nodal Marginal Zone Lymphoma (NMZL), Extranodal Marginal Zone Lymphoma (ENMZL) of Mucosa-Associated Lymphoid Tissue (MALT), Splenic Marginal Zone Lymphoma (SMZL), LymphoPlasmacytic Lymphoma (LPL) and Small Lymphocytic Lymphoma (SLL).
Follicular Lymphoma is the most indolent form and second most common form of all NHLs, and they are a heterogeneous group of lymphoproliferative malignancies. Approximately 20% of all NHLs are Follicular Lymphomas and the average age of diagnosis is 65 years. Advanced stage indolent NHL is not curable and as such, prolonging Progression Free Survival (PFS) and Overall Survival (OS), while maintaining Quality of Life, have been the goals of treatment intervention. Asymptomatic patients with indolent NHL are generally considered candidates for “watch and wait” approach. Patients with advanced stage symptomatic Follicular Lymphoma are often treated with induction chemoimmunotherapy followed by maintenance RITUXAN® (Rituximab). This can result in a median Progression Free Survival (PFS) of 6-8 yrs and a median Overall Survival (OS) of 12-15 yrs. However, approximately 30% of the patients will relapse in 3 years, with prognosis worsening after each subsequent relapse. Despite advances in treatment for Follicular Lymphoma, there remains an unmet need for additional options that offer treatment-free intervals with durable, complete responses.
Loncastuximab tesirine (ZYNLONTA®) is an Antibody-Drug Conjugate (ADC) and consists of a humanized monoclonal antibody (anti-CD19) linked to a cytotoxic alkylating agent (pyrrolobenzodiazepine dimer, or PBD). Upon binding to CD19 on B-calls, Loncastuximab is internalized into the cells and PBD is released inside the cells, where it crosslinks DNA and induces tumor cell death. Loncastuximab is presently indicated for the treatment of adult patients with relapsed or refractory Large B-Cell Lymphoma after two or more lines of systemic therapy, including Diffuse Large B-Cell Lymphoma (DLBCL) not otherwise specified, DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma. Preliminary data suggested promising activity of Loncastuximab in Follicular Lymphoma, with and synergistic activity between Rtuximab-induced cytotoxicity and Loncastuximab.
The researchers in this study evaluated Loncastuximab tesirine combined with Rituximab for second-line and later treatment of Follicular Lymphoma. This single institution, investigator-initiated, Phase 2 trial enrolled 39 patients with Grade 1-3A Relapsed/Refractory Follicular Lymphoma treated with one or more lines of therapy, who presented with progression of disease within 24 months (POD24). Treatment for the initial 21 weeks consisted of Rituximab 375 mg/m2 IV weekly for 4 weeks followed by 1 dose every 8 weeks for a total of 5 doses in combination with Loncastuximab 0.15 mg/kg IV every 3 weeks for 2 doses followed by 0.075 mg/kg IV every 3 weeks for a total of 7 doses. Premedication with Dexamethasone 4 mg twice daily for 3 days was required. Patients achieving Complete Response at Week 21 discontinued Loncastuximab and received two more doses of Rituximab every 8 weeks. Patients with a partial response at week 21 continued both agents for 18 more weeks. No prophylaxis was required per study protocol. The median age of patients was 68 years and majority of patients were men (54%) with advanced-stage (82%), high-disease burden by GELF criteria (92%), and/or POD24 after frontline immunochemotherapy (51%). The median FLIPI score was 3 with most patients assigned to the high-risk group (61.5%). Median lines of prior therapy were 1, and R-CHOP was the most common first-line therapy (56.5%), followed by Bendamustine with Rituximab (25.6%), single-agent Rituximab (15.3%), and Fludarabine, Mitoxantrone, and Dexamethasone (2.6%). The Primary endpoint was Complete Metabolic Response (CMR) rate at week 12, assessed by the Lugano 2014 classification, in patients who had received at least three doses of Loncastuximab. The safety analysis included all patients who received one or more doses of Loncastuximab.
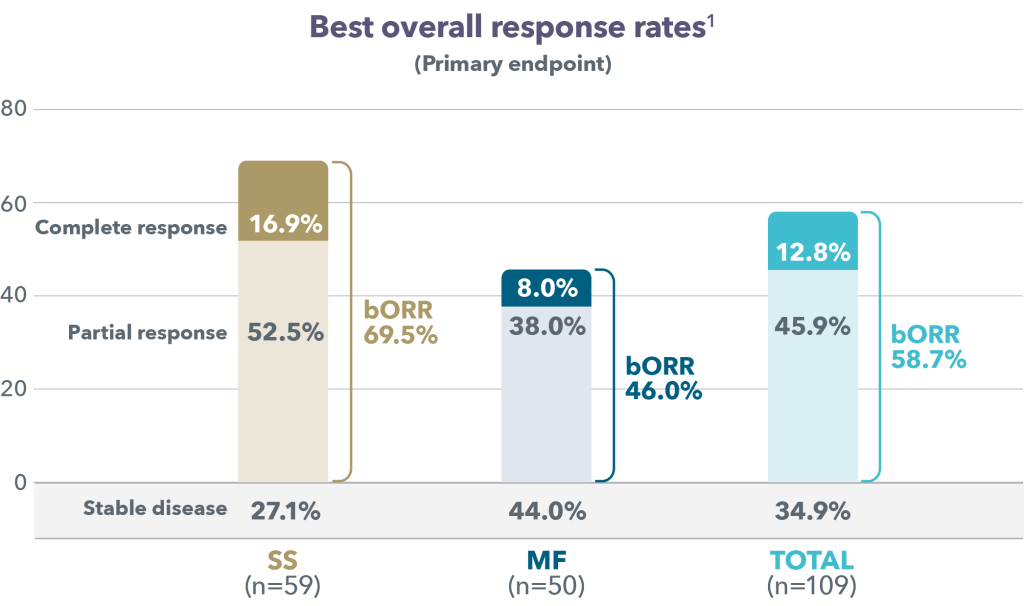
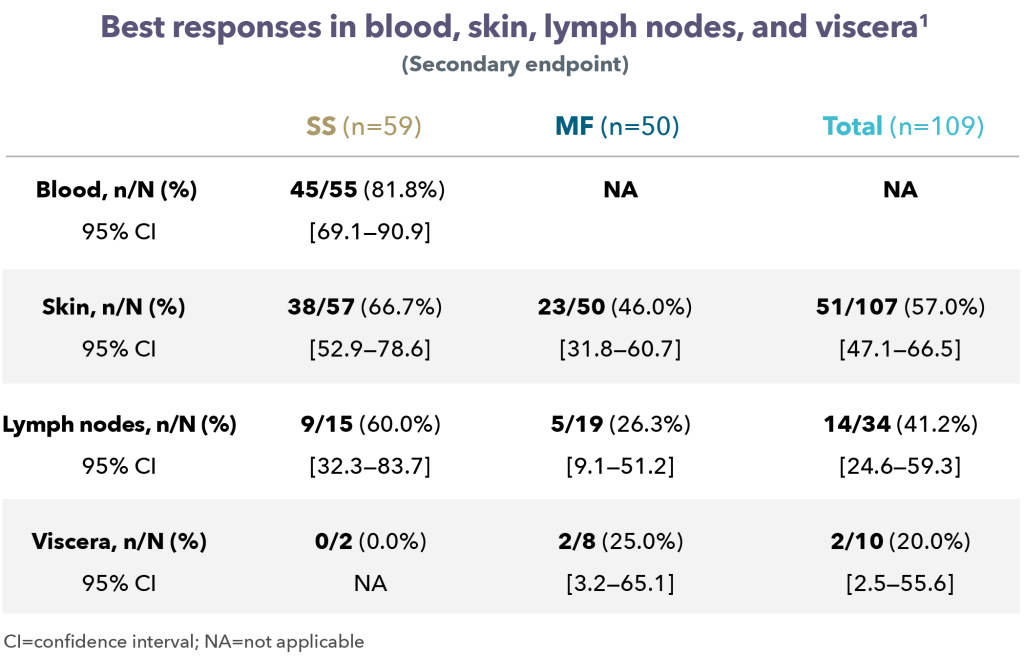
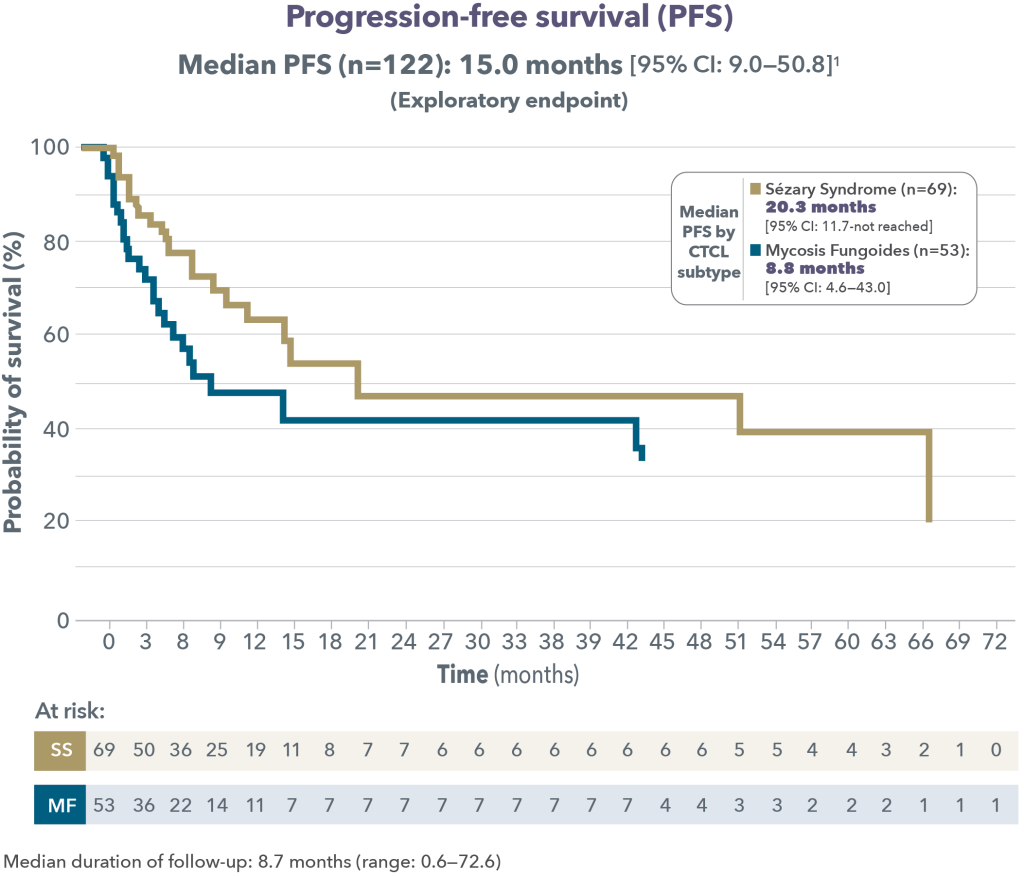
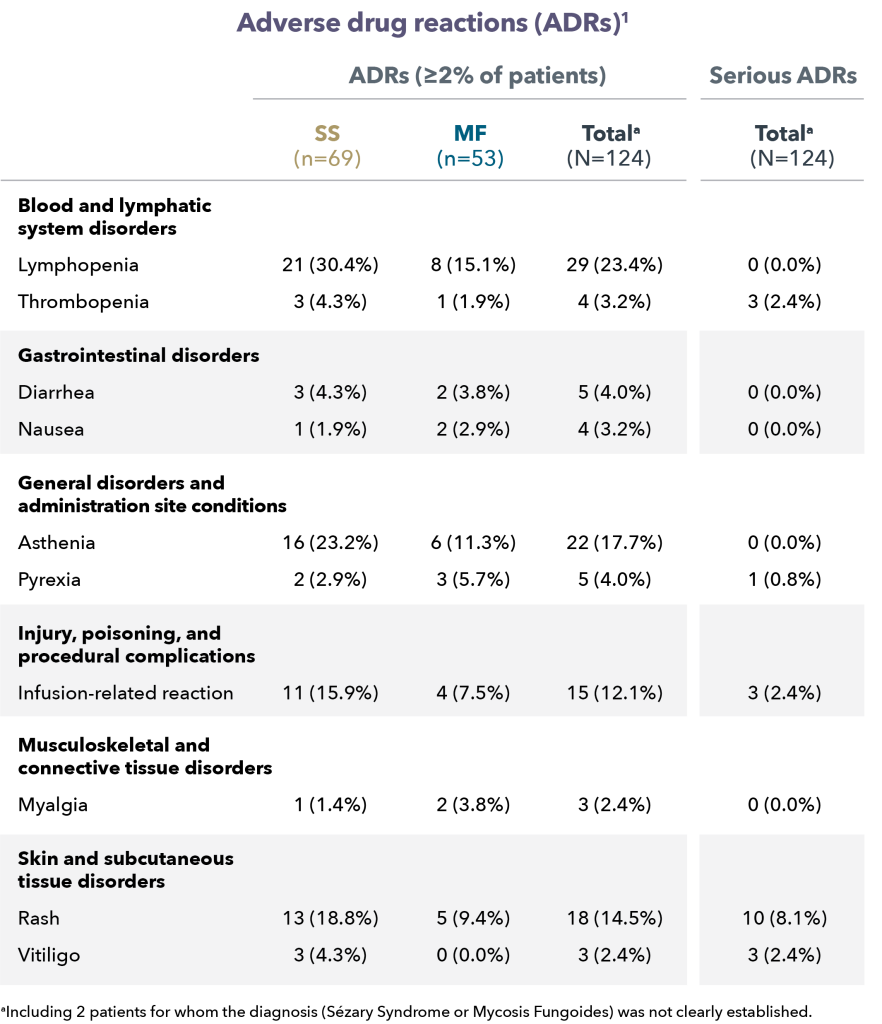
At a median follow-up of 18.2 months, the Objective Response Rate (ORR) at week 12 was 97.1%, with a CMR rate of 68.6% and a Partial Metabolic Response (PMR) rate of 28.6%. The CMR rate improved to 80% at week 21. All patients who achieved a CMR maintained their response. Baseline bone marrow involvement resolved in all patients (N=10) at week 12 reassessment. The response rates were similar in patients with POD24 and those without, each of which made up roughly half the patients in the trial. At 18 months, the Progression Free Survival (PFS) rate was 90.1% and the Overall Survival (OS) rate was 93.3%, and the median PFS and OS was not reached. The main adverse events with this therapy were skin rash that worsened with sun exposure, and fluid retention, which could be treated with diuretics.
In conclusion, a combination of Loncastuximab with Rituximab demonstrated dramatic activity with robust Complete Metabolic Rate and promising survival benefit with manageable toxicities, in patients with high risk Relapsed or Refractory Follicular Lymphoma. The results of this study support this combination as a new treatment option for this patient group, and multicenter expansion cohort is ongoing. Because of the high Complete Response by week 12, the researchers recently reduced the treatment length from 10 to 6 months, with the hope that relatively short course of treatment combined with lower toxicity will allow patients to better tolerate and complete the therapy.
Loncastuximab tesirine with rituximab in patients with relapsed or refractory follicular lymphoma: a single-centre, single-arm, phase 2 trial. Alderuccio JP, Alencar AJ, Schatz JH, et al. The Lancet. 2025;12:E23-E34.