
The FDA on December 18, 2019 granted accelerated approval to PADCEV® for adult patients with locally advanced or metastatic urothelial cancer who have previously received a Programmed Death receptor-1 (PD-1) or Programmed Death-Ligand 1 (PD-L1) inhibitor, and a Platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting. PADCEV® is a product of Astellas Pharma US, Inc.
Tag: Urothelial Cancer (Bladder-Ureters-Renal-Pelvis)
FDA Grants Accelerated Approval to PADCEV® for Metastatic Urothelial Cancer
SUMMARY: The FDA on December 18, 2019, granted accelerated approval to PADCEV® (Enfortumab vedotin-ejfv), for adult patients with locally advanced or metastatic urothelial cancer who have previously received a Programmed Death receptor-1 (PD-1) or Programmed Death-Ligand1 (PD-L1) inhibitor, and a Platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting. The American Cancer Society estimates that in 2019, approximately 80,470 new cases of Bladder Cancer will be diagnosed and 17,670 patients will die of the disease. Patients with urothelial carcinoma are currently treated in the first line setting with a Platinum based chemotherapy regimen and a Check Point Inhibitor (PD-1 or PD-L1 inhibitor) in the second line setting. Treatment options for patients who progress after first and second line therapies are limited, with poor outcomes. The response rates with standard chemotherapy in this patient population, is about 10%.
PADCEV® is an Antibody-Drug Conjugate (ADC) that targets Nectin-4, a cell adhesion molecule highly expressed in urothelial cancers and other solid tumors. Following binding to Nectin-4 on the cell surface, PADCEV® becomes internalized and is processed by lysosomes, with the liberation of its cytotoxic payload, Monomethyl auristatin E, which in turn disrupts microtubule assembly, leading to cell cycle arrest and apoptosis. In a Phase I dose-finding study of PADCEV®, the Objective Response Rate (ORR) was 42% among patients with advanced urothelial cancer, who previously received treatment with a PD-1/PD-L1 inhibitor.
This FDA approval was based on the results from the pivotal Phase II EV-201 study, which is an open-label, single-arm, multicenter trial in which 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and Platinum-based chemotherapy were enrolled. Patients received PADCEV® 1.25 mg/kg on days 1, 8, and 15 of a 28-day cycle, until disease progression or unacceptable toxicity. The median age was 69 years. The Primary endpoint was ORR as assessed by blinded Independent Central Review. Secondary endpoints included Duration of Response, Progression Free Survival (PFS), Overall Survival (OS), Safety and Tolerability.
The ORR was 44%, with 12% Complete Responses and 32% Partial Responses. Overall, 84% of evaluable patients showed some degree of tumor shrinkage. The responses were noted at a median of 1.8 months after treatment initiation and the median Duration of Response was 7.6 months. These objective responses were seen in all patient subgroups evaluated, including those with poor prognostic features. The median PFS was 5.8 months, and the median Overall Survival was 11.7 months. The most common adverse reactions were fatigue, alopecia, decreased appetite and peripheral neuropathy. Blood glucose levels should be monitored closely in patients with, or at risk, for diabetes mellitus or hyperglycemia.
It was concluded from this study that treatment with PADCEV® demonstrated clinically meaningful Objective Response Rate, in patients with advanced metastatic urothelial cancer, who received prior treatment with a PD-1 or PD-L1 inhibitor and Platinum-based chemotherapy, thus fulfilling an unmet need. PADCEV® is the first Nectin-4-directed Antibody-Drug Conjugate to receive FDA approval, and a Phase III study is underway comparing PADCEV® against standard single-agent chemotherapy, in patients with advanced, previously treated metastatic urothelial cancer. EV-201: Results of enfortumab vedotin monotherapy for locally advanced or metastatic urothelial cancer previously treated with platinum and immune checkpoint inhibitors. Petrylak DP, Balar AV, O’Donnell PH, et al. DOI: 10.1200/JCO.2019.37.18_suppl.LBA4505 Journal of Clinical Oncology 37, no. 18_suppl (June 20, 2019) 4505-4505.
FDA Approves BALVERSA® for Metastatic Urothelial Carcinoma
SUMMARY: The FDA on April 12, 2019 granted accelerated approval to BALVERSA® (Erdafitinib) for patients with locally advanced or metastatic Urothelial Carcinoma, with susceptible FGFR3 or FGFR2 genetic alterations,that has progressed during or following platinum-containing chemotherapy, including within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy. Patients should be selected for therapy based on an FDA-approved companion diagnostic for BALVERSA®. The FDA also simultaneously approved the THERASCREEN® FGFR RGQ RT-PCR Kit, developed by QIAGEN, for use as a companion diagnostic for this therapeutic indication.
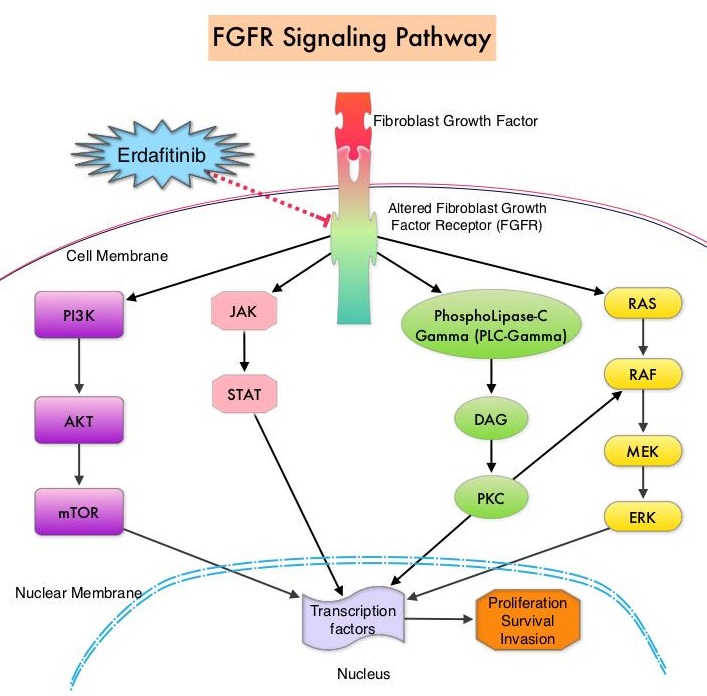 FGFRs are a family of Receptor Tyrosine Kinases, which may be upregulated in a variety of malignancies. The Fibroblast Growth Factor/Fibroblast Growth Factor Receptor (FGF/FGFR) signaling pathway regulates embryogenesis, adult tissue homeostasis, angiogenesis and wound repair, and is also pivotal in cell functions, including proliferation, differentiation, apoptosis and migration. Deregulated FGF/FGFR activations have been associated with developmental disorders and cancer progression. Following binding with a ligand, FGFRs activate downstream signaling pathways such as the Mitogen Activated Protein Kinase (MAPK), Signal Transducer and Activator of Transcription (STAT), the PhosphoInositide-3-Kinase (PI3K)/Akt pathways, and PLC-DAG-PKC pathway. FGFR isoforms have been shown to result in oncogenic FGFR signaling, which in turn promotes tumorigenesis. FGFR3 mutations have been described in approximately 75% of low-grade papillary bladder cancers, and FGFR3 overexpression has been noted in 42% of muscle-invasive bladder cancers. FGFR1 amplification has also been found in 3% of urinary bladder cancers. Patients with FGFR alterations have poor outcomes when treated with available therapies and these alterations occur in 20% of patients with metastatic Urothelial Carcinoma.
FGFRs are a family of Receptor Tyrosine Kinases, which may be upregulated in a variety of malignancies. The Fibroblast Growth Factor/Fibroblast Growth Factor Receptor (FGF/FGFR) signaling pathway regulates embryogenesis, adult tissue homeostasis, angiogenesis and wound repair, and is also pivotal in cell functions, including proliferation, differentiation, apoptosis and migration. Deregulated FGF/FGFR activations have been associated with developmental disorders and cancer progression. Following binding with a ligand, FGFRs activate downstream signaling pathways such as the Mitogen Activated Protein Kinase (MAPK), Signal Transducer and Activator of Transcription (STAT), the PhosphoInositide-3-Kinase (PI3K)/Akt pathways, and PLC-DAG-PKC pathway. FGFR isoforms have been shown to result in oncogenic FGFR signaling, which in turn promotes tumorigenesis. FGFR3 mutations have been described in approximately 75% of low-grade papillary bladder cancers, and FGFR3 overexpression has been noted in 42% of muscle-invasive bladder cancers. FGFR1 amplification has also been found in 3% of urinary bladder cancers. Patients with FGFR alterations have poor outcomes when treated with available therapies and these alterations occur in 20% of patients with metastatic Urothelial Carcinoma.
BALVERSA® (Erdafitinib) is a once-daily, oral, pan-Fibroblast Growth Factor Receptor (FGFR) Tyrosine Kinase Inhibitor. The approval of BALVERSA® was based on data from a cohort of 87 patients, enrolled in Study BLC2001, which is a multicenter, open-label, single-arm trial. Enrolled patients had locally advanced or metastatic Urothelial Carcinoma that had progressed during, or following at least one prior chemotherapy regimen, and had FGFR genomic alterations such as FGFR3 gene mutations or FGFR2 or FGFR3 gene fusions. Ten percent of patients were chemo naïve, 47% percent of patients had received two or more prior lines of therapy and 80% of patients had visceral metastases. Approximately 97% patients had prior Platinum based therapy and 24% of patients had received anti–PD-1/PD-L1 treatment. The median patient age was 67 years. Patients received BALVERSA® at a starting dose of 8 mg PO once daily. Patients whose serum phosphate levels were below the target of 5.5 mg/dL between days 14 and 17 (41% of the patients) had their dose increased to 9 mg once daily. Treatment was continued until disease progression or unacceptable toxicity. The Primary end point was Objective Response Rate (ORR).
The ORR was 32.2%, with Complete Responses in 2.3% and Partial Responses in 29.9%. Median response duration was 5.4 months. Responding patients included those patients who had previously not responded to anti PD-L1 or PD-1 treatment. The most common adverse reactions were increased serum phosphate, stomatitis, fatigue, increased serum creatinine, diarrhea, onycholysis, increased liver function studies and hyponatremia.
The authors concluded that treatment with BALVERSA® resulted in high Response Rates among patients with chemorefractory metastatic Urothelial Carcinoma with FGFR genomic alterations. BALVERSA® is the first approved personalized treatment, targeting susceptible FGFR genetic alterations, fulfilling an unmet need for these poor prognosis patients. First results from the primary analysis population of the phase 2 study of erdafitinib (ERDA; JNJ-42756493) in patients (pts) with metastatic or unresectable urothelial carcinoma (mUC) and FGFR alterations (FGFRalt). Siefker-Radtke AO, Necchi A, Park SH, et al. J Clin Oncol 36, 2018 (suppl; abstr 4503)
BALVERSA® (Erdafitinib)
The FDA on April 12, 2019 granted accelerated approval to BALVERSA® (Erdafitinib) for patients with locally advanced or metastatic Urothelial Carcinoma, with susceptible FGFR3 or FGFR2 genetic alterations,that has progressed during or following Platinum-containing chemotherapy, including within 12 months of neoadjuvant or adjuvant Platinum-containing chemotherapy. Patients should be selected for therapy based on an FDA-approved companion diagnostic for BALVERSA®. The FDA also simultaneously approved the THERASCREEN® FGFR RGQ RT-PCR Kit, developed by QIAGEN, for use as a companion diagnostic for this therapeutic indication. BALVERSA® is a product of Janssen Pharmaceutical Companies.
KEYTRUDA® (Pembrolizumab) and TECENTRIQ® (Atezolizumab)
The FDA on August 16, 2018 updated the prescribing information for these two agents, to require the use of an FDA-approved companion diagnostic test to determine PD-L1 levels in tumor tissue from patients with locally advanced or metastatic urothelial cancer who are Cisplatin-ineligible. FDA approved two different companion diagnostic tests, the Dako PD-L1 IHC 22C3 PharmDx Assay (Dako North America, Inc.) as a companion diagnostic for treatment with KEYTRUDA® and Ventana PD-L1 (SP142) Assay® (Ventana Medical Systems, Inc.) as a companion diagnostic test for treatment with TECENTRIQ®.
KEYTRUDA® Improves Overall Survival in Advanced Urothelial Carcinoma
SUMMARY: The American Cancer Society estimates that in 2017, approximately 79,030 new cases of Bladder Cancer will be diagnosed and 16,870 patients will die of the disease. Patients with urothelial carcinoma are currently treated in the first line setting with a platinum based chemotherapy regimen. Treatment options for patients who progress after platinum based chemotherapy are limited, with poor outcomes. The response rates with standard chemotherapy in this patient population, is about 10%.
The FDA approved KEYTRUDA® (Pembrolizumab) in May 2017 for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy, or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. The FDA subsequently in July 2017 granted an accelerated approval to frontline KEYTRUDA® for patients with locally advanced or metastatic urothelial carcinoma, who are not eligible for cisplatin-containing chemotherapy. KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. By doing so, it unleashes the tumor-specific effector T cells, and is thereby able to undo PD-1 pathway-mediated inhibition of the immune response. 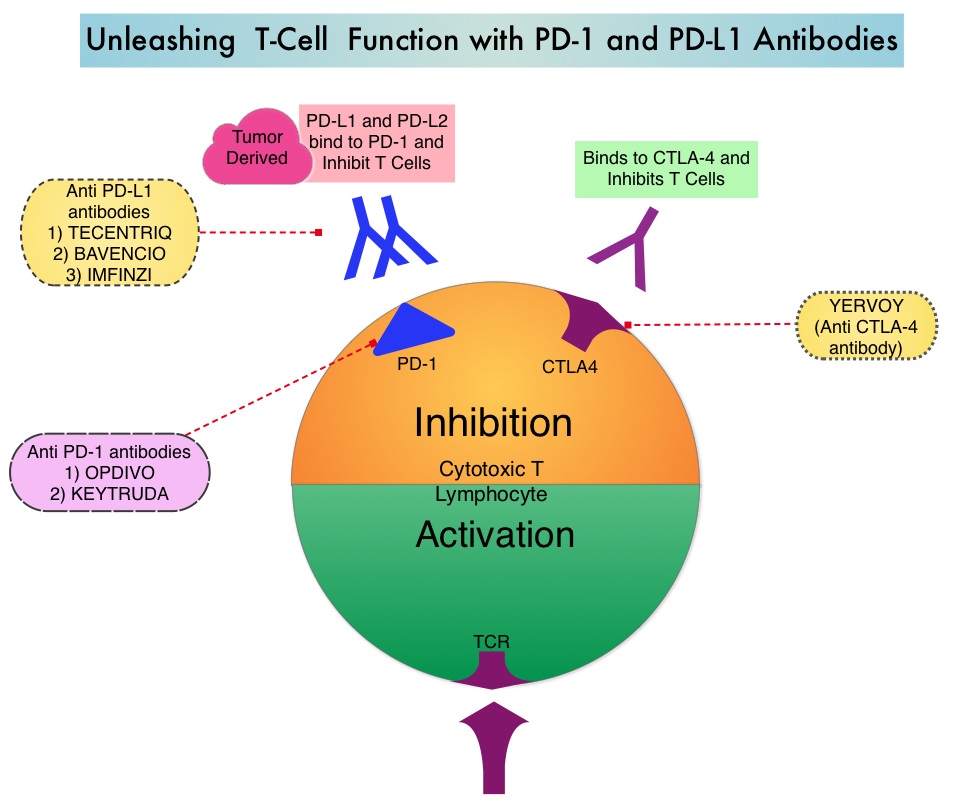
KEYNOTE-045 trial is an open-label, multicenter, phase III study in which 542 patients with advanced urothelial carcinoma who had progressed on prior therapies were randomly assigned to receive KEYTRUDA® or investigator's choice of Paclitaxel, Docetaxel, or Vinflunine. Eligible patients had histologically or cytologically confirmed urothelial carcinoma and had progressed on no more than 2 prior systemic therapies, including a platinum based regimen. Patients were randomly assigned in a 1:1 ratio to receive KEYTRUDA® 200 mg every 3 weeks (N=270) or investigator's choice of Paclitaxel 175 mg/m2 , Docetaxel 75 mg/m2, or Vinflunine 320 mg/m2, every 3 weeks (N=272). The primary endpoints were Overall Survival (OS) and Progression Free Survival (PFS), and the secondary endpoints included Objective Response Rate (ORR) and Safety. Efficacy was assessed in all patients as well as in patients with a PD-L1 Combined Positive Score (CPS) of 10% or more. (CPS is the percentage of PD-L1-expressing tumor and inflammatory cells).
In an updated analysis, with a median follow up of 22.5 months for both treatment groups, the median OS with KEYTRUDA® was 10.3 months compared with 7.4 months with chemotherapy (HR=0.70; P=0.0003) and among patients with a Combined Positive Score of 10% or more, the OS was 8.0 versus 5.2 months respectively (HR=0.58; P=0.003). The OS benefit was noted regardless of age, liver metastases, hemoglobin, visceral disease, and choice of chemotherapy. The 18 month OS rate was 33.2% with KEYTRUDA® versus 19.7% with chemotherapy. There was however no significant difference in the median PFS between the two treatment groups. The ORR was 21.1% with KEYTRUDA® and 11.0% with chemotherapy and the responses with KEYTRUDA® were more durable than with chemotherapy. The median response duration of response was not reached in the KEYTRUDA® group versus 4.4 months in the chemotherapy group. Treatment-related Adverse Events of any grade occurred in 62% of patients in the KEYTRUDA® group and 91% of patients in the chemotherapy group. Discontinuation due to toxicities occurred in 7.1% versus 12.5% of KEYTRUDA® vs chemotherapy patients, respectively.
It was concluded that KEYTRUDA® is the first agent to improve Overall Survival over chemotherapy, in the second line setting, for patients with recurrent, advanced urothelial carcinoma, and a significant proportion of patients who respond, have very durable responses. Pembrolizumab (pembro) versus paclitaxel, docetaxel, or vinflunine for recurrent, advanced urothelial cancer (UC): mature results from the phase 3 KEYNOTE-045 trial. De Wit R, Vaughn DJ, Fradet Y, et al. Annals of Oncology (2017) 28 (suppl_5): v605-v649. 10.1093/annonc/mdx440
E-Cigarettes Can Increase the Risk of Bladder Cancer
SUMMARY: The American Cancer Society estimates that tobacco use is responsible for nearly 1 in 5 deaths in the United States and accounts for at least 30% of all cancer deaths. Smokeless tobacco products are a major source of cancer causing nitrosamines, and increase the risk of developing cancer of the oropharynx, esophagus, and pancreas. Cigarette smoke contains more than 7,000 chemicals, many of which are toxic and some linked to cancer. E-cigarettes or Electronic Nicotine Delivery Systems (ENDS) were first developed in China and introduced to the U.S. market in 2007. When a smoker inhales through the mouth piece of an e-cigarette, the air flow triggers a sensor that switches on a small lithium battery powered heater, which in turn vaporizes liquid nicotine along with PolyEthylene Glycol (PEG) present in a small cartridge. The PEG vapor looks like smoke. The potent liquid form of nicotine extracted from tobacco is tinctured with fragrant flavors such as chocolate, cherry and bubble gum, coloring substances, as well other chemicals and these e-liquids are powerful neurotoxins.
With the rapid growth of the e-cigarette industry and the evidence of potential dangers and risk to public health, particularly children, experts from the world's leading lung organizations were compelled to release a position statement on electronic cigarettes, specifically focusing on their potential adverse effects on human health and calling on government organizations to ban or restrict the use of e-cigarettes, until their impact on health is better understood. According to the National Youth Tobacco Survey, the use of e-cigarettes has tripled from 2013 to 2014 among middle school and high school students. Epidemiological data have shown that nicotine use is a gateway to the use of cocaine and marijuana and subsequent lifelong addiction. E-cigarettes and other Electronic Nicotine Delivery Systems (ENDS), unlike combustible cigarettes and many other tobacco products are not currently regulated by the U.S. Food and Drug Administration.
Smoking more than one pack of cigarettes a day has been associated with an increased risk of mortality among patients with bladder cancer. E-cigarettes have been advertised and promoted as a safer way of delivering the stimulating effects of tobacco smoke, without the harmful health risks. Contrary to this claim, over 90% of inhaled nicotine is excreted to urine. Nicotine can be nitrosatized in urothelial cells and then further metabolized into carcinogenic nitrosamines and formaldehyde, which in turn can induce DNA damage in the bladder mucosa. Additionally, these metabolites can also block DNA repair, increasing cancer risk.
The authors in this study compared the urine of e-cigarette users to that of nonsmokers, for known bladder carcinogens. There are five known bladder carcinogens that are either present in traditional cigarettes or common solvents believed to be used in some e-cigarette formulations, and they include (benz(a)anthracene, benzo(a)pyrene, 1-hydroxypyrene, o-toluidine and 2-naphthylamine. The limit of detection of these carcinogens in this study was 10-100 ng/ml. Urine samples were collected from 13 e-cigarette users and 10 non-smoking, non e-cigarette using controls. The mean age was 39 years and participants were predominantly male and had abstained completely from traditional cigarettes, for at least 6 months prior to specimen collection.
It was noted that the urine from 92% of e-cigarette users tested positive for two of the five carcinogenic compounds whereas none of the controls tested positive for these carcinogens. Interestingly, all study participants considered e-cigarettes were safe, when interviewed by the study investigators.
It was concluded that in this important pilot study, majority of the e-cigarette users had carcinogenic metabolites in the urine, which can contribute to the development of bladder cancer. This study underscores the importance both traditional and e-cigarette smoking cessation, for bladder cancer prevention. MP88-14 EVALUATION OF E-CIGARETTES USERS URINE FOR KNOWN BLADDER CARCINOGENS. Fuller T, Acharya A, Bhaskar G, et al. DOI: http://dx.doi.org/10.1016/j.juro.2017.02.2739
KEYTRUDA® (Pembrolizumab)
The FDA on May 18, 2017 granted regular approval to KEYTRUDA® for patients with locally advanced or metastatic Urothelial Carcinoma, who have disease progression during or following Platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with Platinum-containing chemotherapy. KEYTRUDA® is a product of Merck and Co., Inc.
FDA Approves IMFINZI® for Advanced Bladder Cancer
SUMMARY: The FDA on May 1, 2017, granted accelerated approval to IMFINZI® (Durvalumab) for the treatment of patients with locally advanced or metastatic Urothelial Carcinoma who have disease progression during or following platinum-containing chemotherapy or who have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. The FDA also approved the VENTANA PD-L1 (SP263) Assay as a complementary diagnostic for the assessment of the PD-L1 protein in Formalin-Fixed, Paraffin-Embedded Urothelial Carcinoma tissue. Urothelial Carcinoma accounts for 90% of all bladder cancers and can originate in the renal pelvis, ureter and urethra. The American Cancer Society estimates that in 2017, approximately 79,030 new cases of Bladder Cancer will be diagnosed and 16,870 patients will die of the disease. Treatment options for patients who progress after platinum based chemotherapy are limited, with poor outcomes. The response rates with standard chemotherapy in this patient population, is about 10%.
The treatment paradigm for solid tumors has been rapidly evolving, with a better understanding of the Immune checkpoints or gate keepers. Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), as well as Programmed cell Death Ligands (PD-L1) that are expressed by cells in the tumor micro environment. By inhibiting checkpoint proteins and their ligands, T cells are unleashed, resulting in T cell proliferation, activation and a therapeutic response. It has been noted that PD-L1 is widely expressed in tumor and immune cells of patients with Urothelial Carcinoma. This in turn helps cancer cells to evade detection from the immune system by binding to the PD-1 receptor on cytotoxic T lymphocytes.
IMFINZI® is a selective, high-affinity human IgG1 monoclonal antibody directed against PD-L1 and blocks the interaction of PD-L1 with PD-1 and CD80. The accelerated FDA approval of IMFINZI® was based on evaluation of 182 patients in the bladder cancer cohort of Study 1108, which is a single-arm Phase I/II trial. This study evaluated the safety and efficacy of IMFINZI® in patients with locally-advanced or metastatic Urothelial Carcinoma of the bladder, who had progressed while on or after a platinum-containing chemotherapy regimen. This study included patients who had progressed within 12 months of receiving therapy, in a neoadjuvant or adjuvant setting. Treatment consisted of IMFINZI® 10 mg/kg IV every 2 weeks until disease progression or unacceptable toxicity. Tumor PD-L1 expression was assessed using the validated VENTANA SP263 Assay, in Formalin-Fixed, Paraffin-Embedded Urothelial Carcinoma tissue. High PD-L1 expression was defined as 25% or more expression on tumor and immune cells. The Primary endpoints were Objective Response Rate and Safety. Secondary endpoints included Duration of Response (DoR) and Overall Survival (OS).
The confirmed Objective Response Rate as assessed by blinded Independent Central Review was 17% and the median response duration was not reached. Objective Response Rate was also analyzed by PD-L1 expression status. In the 182 patients, the confirmed Objective Response Rate was 26.3% in patients with a high PD-L1 score and 4.1% in patients with a low or negative PD-L1 score. The most common adverse reactions were fatigue, musculoskeletal pain, constipation, decreased appetite, nausea, peripheral edema, and urinary tract infection.
The authors concluded that IMFINZI® has significant clinical activity and an excellent safety profile in patients with locally advanced or metastatic Urothelial Carcinoma. Clinical trials are underway, evaluating IMFINZI® as monotherapy and in combination with other checkpoint inhibitors, in the first line treatment of patients with advanced bladder cancer. Updated Efficacy and Tolerability of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma. Powles T, O’Donnell PH, Massard C, et al. J Clin Oncol. 2017;35 (suppl 6S; abstract 286).
BAVENCIO® (Avelumab)
The FDA on May 9, 2017 granted accelerated approval to BAVENCIO® for patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy. BAVENCIO® is a product of EMD Serono, Inc.