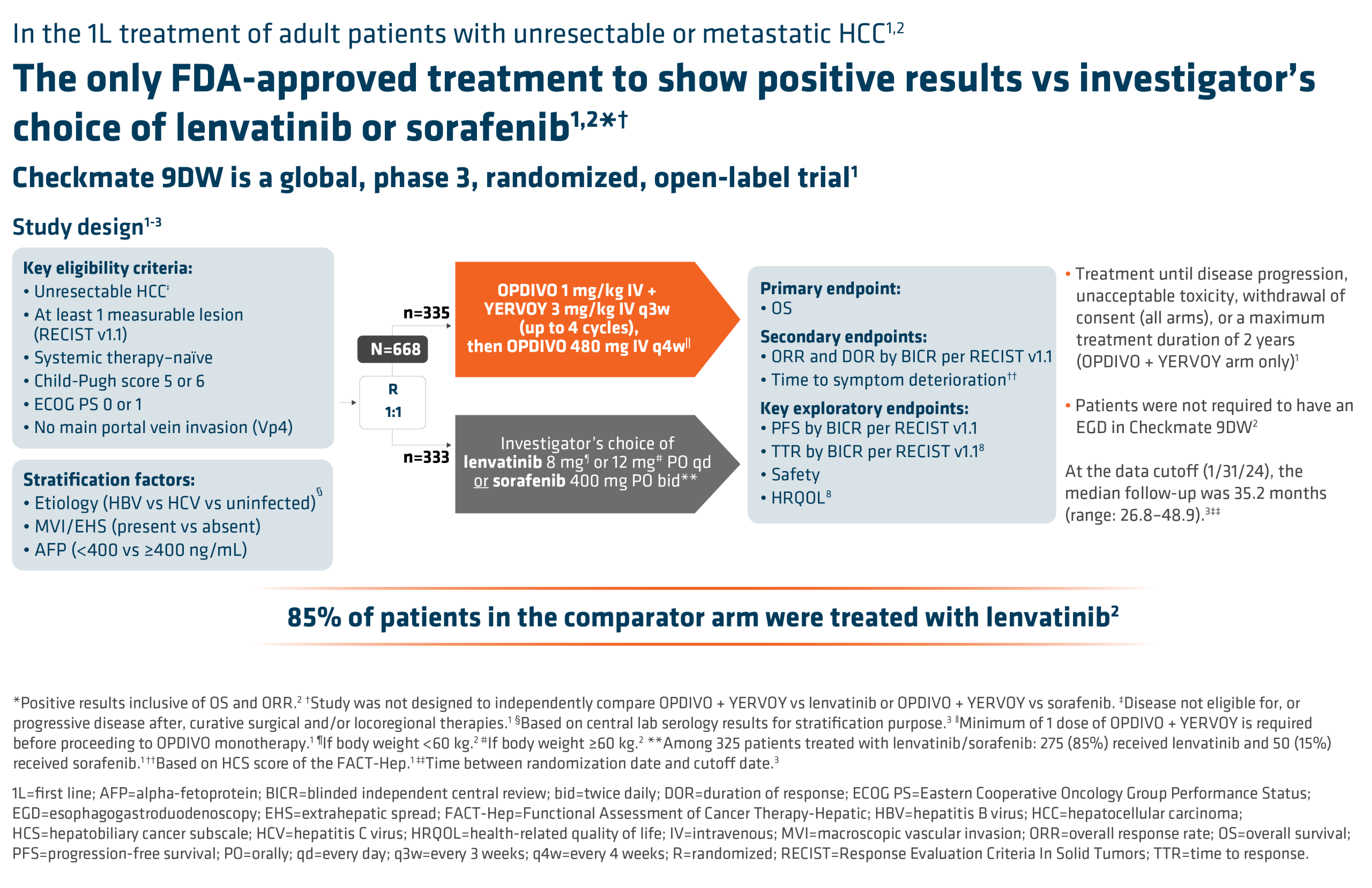
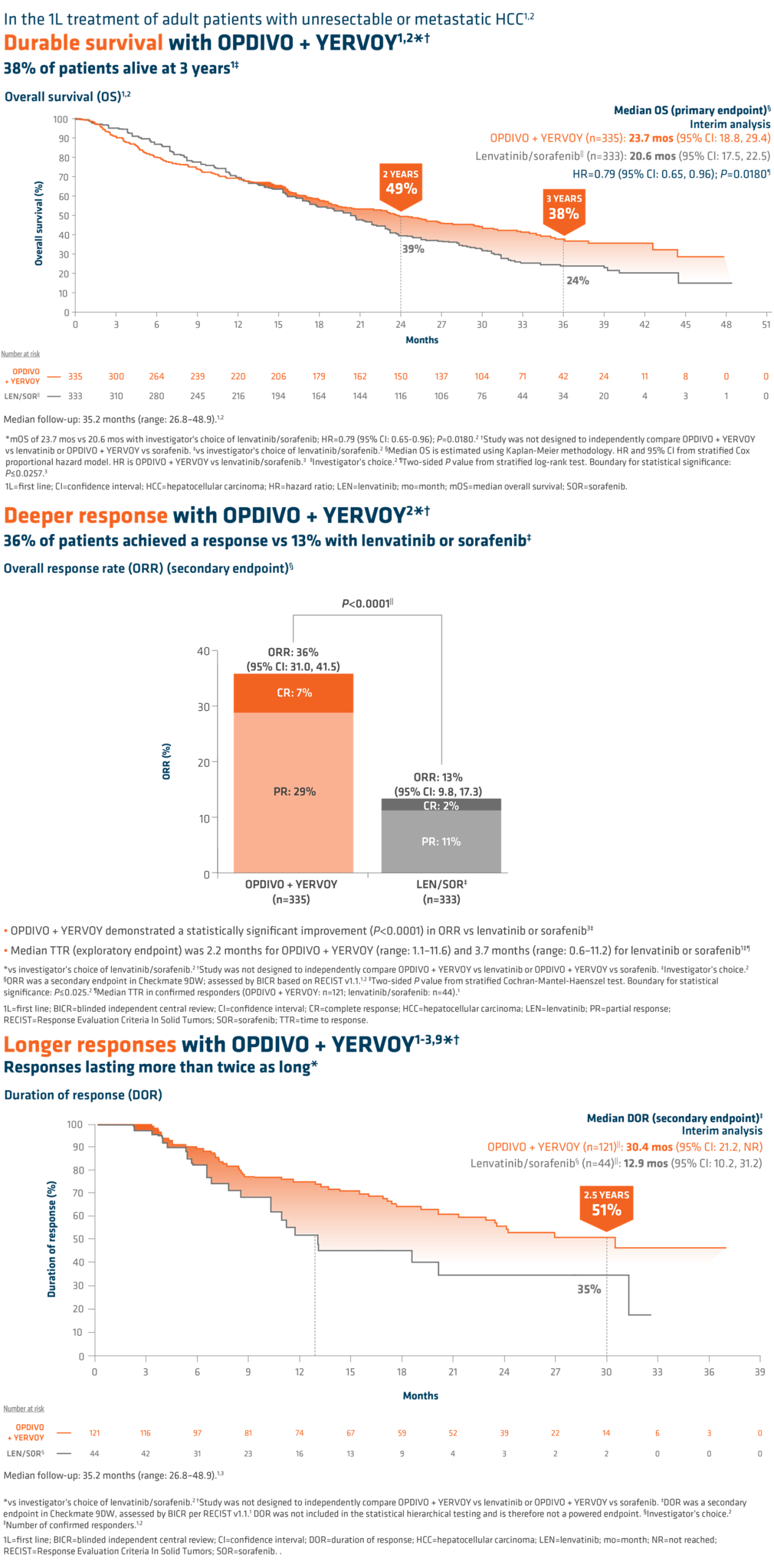
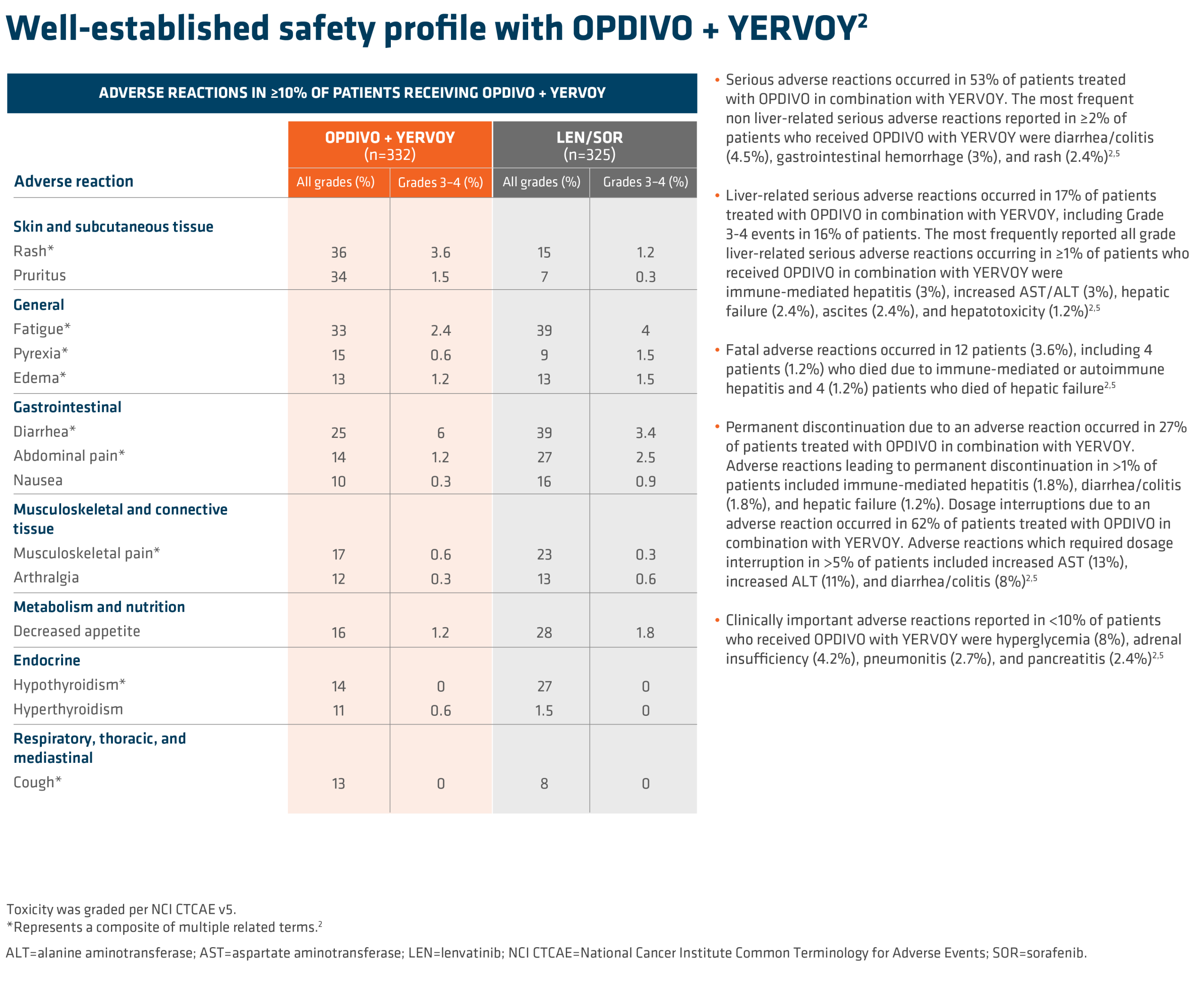
SUMMARY: It is estimated that in the US approximately 13,000 people are diagnosed with MyeloDysplastic Syndromes (MDS) each year. The prevalence has been estimated to be from 60,000 to 170,000 in the US. MyeloDysplastic Syndromes are a heterogenous group of stem cell disorders characterized by marrow failure resulting in cytopenias, mainly symptomatic anemia, with associated cytogenetic abnormalities, and abnormal cellular maturation with morphologic changes in clonal cells. Majority of the individuals diagnosed with MDS are 65 years or older and die as a result of infection and/or bleeding, consequent to bone marrow failure. About a third of patients with MDS develop Acute Myeloid Leukemia (AML).
The International Prognostic Scoring System (IPSS) for MDS has 4 risk groups based on Total Risk Score (Low, Intermediate-1, Intermediate-2 and High). The three prognostic factors scored to predict the course of the patient’s disease include, percentage of blast cells in the bone marrow, type of chromosomal changes in the marrow cells and number of cytopenias (anemia, neutropenia or thrombocytopenia). Patients with low-risk MDS have an indolent disease course with a median survival of about 6 years with no therapeutic intervention. Patients with intermediate and higher-risk disease however have a shorter median survival even with treatment, with approximately a third of the patients progressing to AML within 3 years.
Patients with Low-risk MDS often present with symptomatic anemia and these patients are in chronic need for RBC transfusions. These patients are treated with Erythropoiesis Stimulating Agents (ESAs) as first line therapy. ESAs such as Darbepoetin alfa and Epoetin alfa are re-engineered and recombinant DNA technology products of Erythropoietin (EPO), and they stimulate erythropoiesis by binding and activating the EPO receptor. However, transfusion-dependent patients with serum EPO levels above 200U per liter are less likely to respond to ESAs. A majority of patients with higher-risk MDS are treated with hypomethylating agents such as Azacitidine and Decitabine and these agents can favorably modify the natural history of the disease, and have been shown to improve survival.
Despite the availability of hypomethylating agents (HMAs) for nearly two decades, survival outcomes for patients with high-risk myelodysplastic syndromes (MDS) in the United States have remained largely unchanged. New population-level data from a large Medicare-based analysis shed light on real-world utilization patterns, revealing substantial underuse and deviations from evidence-based treatment practices that may help explain the persistent gap between clinical trial efficacy and real-world effectiveness.
Clinical Context
For patients with MDS, allogeneic hematopoietic stem cell transplantation remains the only curative approach. However, most individuals are ineligible for transplant because of advanced age, comorbidities, or limited donor availability, underscoring the importance of effective pharmacologic therapies. Azacitidine and Decitabine, both FDA-approved HMAs, are the cornerstone of therapy for higher-risk disease. In pivotal trials, Azacitidine improved Overall Survival by approximately 9 months compared with conventional care regimens. Yet, real-world studies have shown little to no improvement in survival outcomes among patients with high-risk MDS over the past two decades.
Study Design and Population
Investigators conducted a retrospective cohort analysis using national Medicare claims data from 2011–2014 to explore factors associated with HMA use in clinical practice. The study identified 49,514 individuals aged ≥65 years diagnosed with incident MDS between 2012 and 2013 (median age 76 years; 53.9% male; 88.4% White). Demographic, clinical, and socioeconomic variables were analyzed to determine predictors of HMA receipt and treatment duration.
Only 16.1% of patients received HMA therapy, despite an estimated 30–40% of newly diagnosed cases representing higher-risk disease for which such therapy is indicated. Of those treated, 73% received Azacitidine, while 27% received Decitabine.
Disparities in HMA Utilization
Multivariable regression analyses revealed striking disparities across age, sex, and race. Compared with patients aged 65–74 years, those aged 75–84 years and ≥85 years had progressively lower odds of receiving HMAs (adjusted odds ratios [aOR], 0.81 and 0.41, respectively). Women were less likely than men to receive treatment (aOR, 0.81), and Black patients were significantly less likely than White patients (aOR, 0.70). These trends suggest potential access barriers, physician bias, or perceived differences in disease risk that may contribute to inequitable care delivery.
Suboptimal Treatment Duration and Adherence
Even among those initiated on therapy, adherence to recommended dosing and duration was poor. Nearly one-third of patients discontinued treatment after a single cycle, and by cycle six, up to half had stopped therapy. Only about half of patients completed the minimum four treatment cycles required to assess efficacy, with even fewer reaching six cycles. Inadequate dosing was also frequent, with 31% of patients failing to complete a full first cycle, and this proportion rose to 40% by the sixth cycle.
These findings stand in sharp contrast to clinical trial protocols, where patients received continuous therapy for at least 4–6 months before response assessment. Premature discontinuation, often due to early cytopenias, perceived lack of benefit, or logistical barriers such as weekend clinic closures, may deprive patients of therapeutic benefit.
Predictors of Shorter Treatment Duration
Factors associated with receipt of fewer than four HMA cycles included treatment with Decitabine (aOR, 0.70), presence of multiple cytopenias (aOR, 0.69), residence in a nursing home (aOR, 0.64), and high frailty scores (aOR, 0.50). These observations highlight how clinical frailty, comorbidity burden, and treatment logistics intersect to affect real-world delivery of care.
Clinical and System-Level Implications
The study underscores two major issues in MDS management: underutilization of effective therapies and nonadherence to evidence-based treatment schedules. Together, these gaps may explain why survival outcomes have not mirrored those seen in pivotal trials. The findings also raise concerns about potential inequities in care, with older, female, and non-White patients less likely to receive HMAs.
Real-world challenges, such as treatment-related cytopenias, patient fatigue, and logistical burdens associated with frequent clinic visits, likely contribute to early discontinuation. Furthermore, variable provider familiarity with MDS treatment guidelines and limited access to pathologic and genomic expertise in community settings may exacerbate these gaps.
Expert Perspective
As study investigators noted, clinical response to HMAs often requires patience and persistence: “Things tend to get worse before they get better.” Awareness of expected early cytopenias and reassurance that delayed response is typical are essential to avoid premature discontinuation. Provider education and patient counseling can play critical roles in improving adherence to treatment duration recommendations.
The authors also emphasize the potential value of consultation at specialized MDS centers or centers of excellence, even if only once at diagnosis. Such consultations can facilitate individualized treatment planning, ensure proper risk stratification, and enable shared-care models with community oncologists, approaches that have been associated with improved outcomes in rare hematologic malignancies.
Conclusion
This large, population-based study exposes significant real-world deficiencies in the use of hypomethylating agents for MDS, both in treatment initiation and duration. Despite clear clinical guidelines and two decades of experience with these agents, underuse, early discontinuation, and inequities in care persist. Strengthening adherence to guideline-based therapy, expanding access to specialized expertise, and addressing system-level barriers may help realize the survival benefits long demonstrated in clinical trials.
Key Takeaways
- Underuse of HMAs: Only 16% of older adults with MDS received hypomethylating agents, despite 30–40% having high-risk disease warranting treatment.
- Disparities in Care: Older age, female sex, and Black race were independently associated with lower odds of receiving HMA therapy.
- Suboptimal Treatment Duration: Nearly half of patients discontinued therapy before completing the minimum 4–6 recommended cycles, often due to early cytopenias or logistical barriers.
- Practice Implications: Improved adherence to guideline-based therapy, enhanced education for community clinicians, and access to expert consultation at MDS specialty centers could help narrow the survival gap between clinical trials and real-world outcomes.
Disparities in Real-World Treatment Patterns of Hypomethylating Agents Among Patients with Myelodysplastic Syndromes in the US. Mukherjee S, Dong W, Gerds AT, et al. Blood Neoplasia. 2025;doi:10.1016/j.bneo.2025.100156.