
The FDA on June 8, 2018 granted regular approval to VENCLEXTA® for patients with Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL), with or without 17p deletion, who have received at least one prior therapy. VENCLEXTA® is a product of AbbVie Inc. and Genentech Inc.
Author: RR
MIRCERA® (Methoxy polyethylene glycol-epoetin beta)
The FDA on June 7, 2018 approved MIRCERA® for the treatment of pediatric patients 5 to 17 years of age on hemodialysis who are converting from another ESA after their hemoglobin level was stabilized with an ESA. MIRCERA® is a product of Vifor Pharma Inc.
FULPHILA® (Pegfilgrastim-jmdb)
The FDA on June 4, 2018 approved FULPHILA® as a biosimilar to NEULASTA® (Pegfilgrastim, Amgen, Inc.), to decrease the chance of infection as suggested by febrile neutropenia in patients with non-myeloid cancer, who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia. FULPHILA® is a product of Mylan GmbH.
DOPTELET® (Avatrombopag)
The FDA on May 21, 2018 approved DOPTELET® for thrombocytopenia in adults with chronic liver disease scheduled to undergo a procedure. DOPTELET® is a product of AkaRx Inc.
RETACRIT® (Epoetin alfa-epbx)
The FDA on May 15, 2018 approved RETACRIT® as a biosimilar to EPOGEN®/PROCRIT® (Epoetin alfa, Amgen Inc.), for the treatment of anemia due to Chronic Kidney Disease (CKD) in patients on dialysis and not on dialysis, use of Zidovudine in patients with HIV infection and the effects of concomitant myelosuppressive chemotherapy. It is also approved for the reduction of allogeneic red blood cell transfusions in patients undergoing elective, noncardiac, nonvascular surgery. RETACRIT® is a product of Hospira Inc., a subsidiary of Pfizer Inc.
Late Breaking Abstract – ASCO 2018 First-Line KEYTRUDA® Superior to Chemotherapy in NSCLC
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2018 about 234,030 new cases of lung cancer will be diagnosed and over 154,050 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells. High level of Programmed Death-Ligand 1 (PD-L1) expression is defined as membranous PD-L1 expression on at least 50% of the tumor cells, regardless of the staining intensity. It is estimated that based on observations from previous studies, approximately 25% of the patients with advanced NSCLC have a high level of PD-L1 expression and high level of PD-L1 expression has been associated with significantly increased response rates to KEYTRUDA®. The FDA approved KEYTRUDA® for the first-line treatment of advanced NSCLC with high PD-L1 expression (Tumor Proportion Score of 50% or more), as well as in combination with Pemetrexed and Carboplatin, as first-line treatment of patients with metastatic nonsquamous NSCLC and for previously treated advanced NSCLC with a PD-L1 Tumor Proportion Score of 1% or more. Currently, KEYTRUDA® is the only FDA approved immunotherapy for initial treatment of NSCLC as monotherapy (KEYNOTE-024) or in combination with chemotherapy. In KEYNOTE-024, KEYTRUDA® significantly improved Progression Free Survival and Overall Survival compared to chemotherapy, as first-line treatment for metastatic NSCLC, without targetable mutations and PD-L1 TPS of 50% or more. KEYNOTE-042 trial evaluated the benefit of KEYTRUDA® in patients whose tumors had a much lower level of PD-L1 expression (TPS of 1% or higher).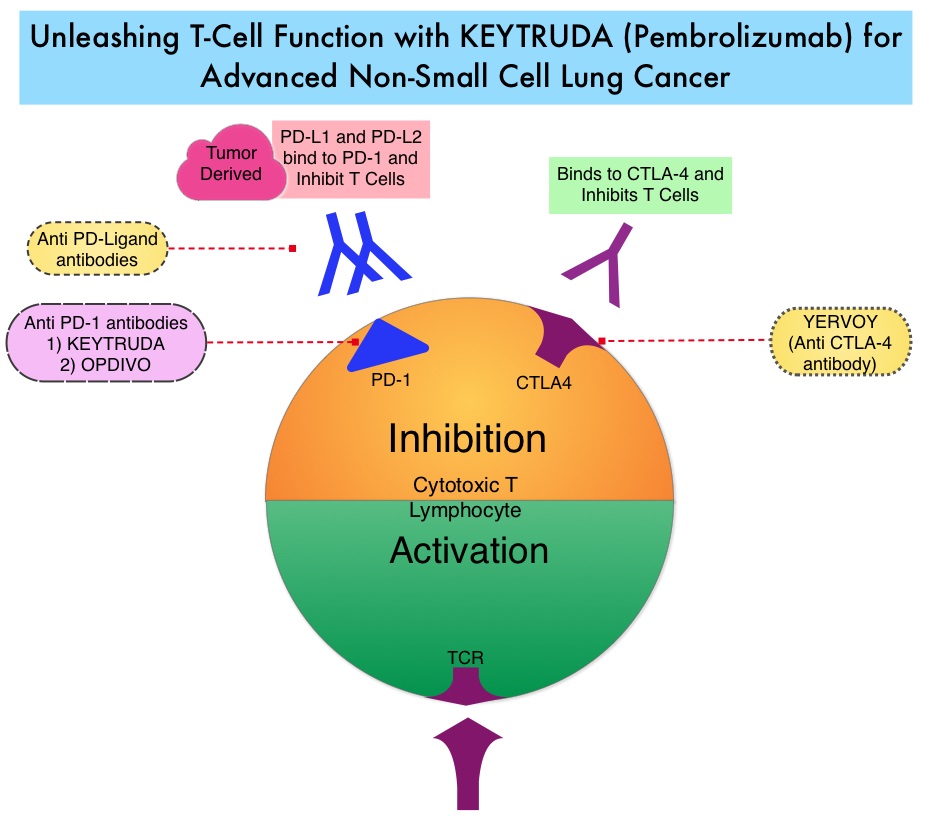
KEYNOTE-042 is a large, international, multicenter, randomized phase III trial in which 1274 patients with untreated locally advanced or metastatic NSCLC were randomly assigned to KEYTRUDA® or chemotherapy with Paclitaxel plus Carboplatin or Pemetrexed plus Carboplatin. In this study, both squamous and non-squamous cancers with PD-L1 Tumor Proportion Score (TPS) of 1% or more were included, but tumors with sensitizing Epidermal Growth Factor Receptor (EGFR) or Anaplastic Lymphoma Kinase (ALK) mutations cancers with genetic changes, that could be treated with targeted therapies such as EGFR and ALK inhibitors, were excluded. Eligible patients were randomly assigned in a 1:1 to receive either KEYTRUDA® 200 mg IV every 3 weeks for up to 35 cycles or investigator’s choice of up to 6 cycles of chemotherapy with Paclitaxel plus Carboplatin or Pemetrexed plus Carboplatin, with optional Pemetrexed maintenance for nonsquamous NSCLC. Patients were divided into 3 treatment groups based on their PD-L1 Tumor Proportion Score (TPS): TPS 50% or more (N=599), TPS 20% or more (N=818), and TPS 1% or more (N=1274). Each PD-L1 expression group had equal numbers of patients receiving KEYTRUDA® and chemotherapy. The Primary end points were Overall Survival (OS) in patients with TPS 50% or more, 20% or more, and 1% or more.
At a median follow up of 12.8 months, 13.7% of patients were still receiving KEYTRUDA® compared with 4.9% on Pemetrexed maintenance therapy. It was noted that KEYTRUDA® was significantly superior to chemotherapy in all PD-L1 expression subsets. In patients with a PD-L1 TPS 50% or more, the median OS with KEYTRUDA® was 20 months versus 12.2 months for chemotherapy (HR=0.69, P=0.0003), for patients with PD-L1 TPS 20% or more, the median OS was 17.7 months versus 13 months respectively (HR=0.77, P=0.002), and for those with PD-L1 TPS 1% or more, the median OS was 16.7 months versus 12.1 months respectively (HR=0.81, P = 0.0018). The Response Rates (RR) were also higher among patients who received KEYTRUDA®, with RR of 39.5% for KEYTRUDA® versus 32% for chemotherapy in patients with a TPS 50% or more, 33.4% and 28.9% respectively in patients with TPS 20% or more and 27.3% and 26.5%, respectively, in patients with TPS of 1% or more. The duration of response was also superior with KEYTRUDA® in all three PD-L1 subgroups compared to chemotherapy (20.2 months versus 8-11 months). Patients receiving KEYTRUDA® experienced fewer severe Adverse Events, compared with chemotherapy (17.8% versus 41%).
The authors concluded that this is the largest clinical trial of KEYTRUDA® as a stand-alone therapy, and is the first study with a Primary end point of OS to demonstrate superiority of KEYTRUDA® over platinum-based chemotherapy, in patients with previously untreated advanced/metastatic NSCLC, without sensitizing EGFR or ALK alterations and a PD-L1 TPS of 1% or more. These data confirmed the benefit of KEYTRUDA® monotherapy as a standard first-line treatment, for PD-L1-expressing advanced/metastatic NSCLC. Pembrolizumab (pembro) versus platinum-based chemotherapy (chemo) as first-line therapy for advanced/metastatic NSCLC with a PD-L1 tumor proportion score (TPS) ≥ 1%: Open-label, phase 3 KEYNOTE-042 study. Lopes G, Wu Y-L, Kudaba I, et al. J Clin Oncol 36, 2018 (suppl; abstr LBA4)
Late Breaking Abstract – ASCO 2018 mFOLFIRINOX Regimen Significantly Improves Overall Survival in Resected Pancreatic Cancer
SUMMARY: The American Cancer Society estimates that in 2018, about 55,440 people will be diagnosed with pancreatic cancer and about 44,330 people will die of the disease. Pancreatic cancer is the fourth most common cause of cancer-related deaths in the United States and Western Europe. Curative surgical resection has been shown to significantly improve Overall Survival (OS) when compared to Chemoradiation, for resectable Pancreatic Cancer. The standard surgical procedure for tumors of the Pancreatic head is the Pancreaticoduodenectomy (Whipple procedure), whereas distal Pancreatectomy is performed for tumors of the body or tail of the Pancreas. Previously published studies concluded that 6 months of Gemcitabine based adjuvant therapy improves Overall Survival for patients with resectable Pancreatic Cancer. FOLFIRINOX chemotherapy regimen however, is more effective than Gemcitabine as first-line treatment, in metastatic pancreatic cancer, for patients with good Performance Status. The following study was conducted to assess the benefit of mFOLFIRINOX regimen in the adjuvant setting.
PRODIGE 24/CCTG PA.6 is a phase III multicenter, randomized clinical trial in which 493 patients were enrolled. Eligible patients had histologically proven, nonmetastatic, pancreatic ductal adenocarcinomas, and had undergone R0 (curative resection) or R1(microscopic residual tumor/positive margins) resection, with no residual tumor on a postoperative CT scan. Patients had a WHO Performance Status of 1 or less and were randomized in a 1:1 ratio, 3-12 weeks after surgery, to receive Gemcitabine on days 1, 8, and 15 every 28 days for 6 cycles (Group A, N=246)) or mFOLFIRINOX regimen, which consisted of Oxaliplatin 85 mg/m², Leucovorin 400 mg/m², Irinotecan 150 mg/m² D1, and 5-FU 2400mg/m² over 46 hours, all drugs given IV, every 14 days for 12 cycles (Group B, N=247). The Primary endpoint was Disease Free Survival (DFS) and Secondary endpoints included Overall Survival (OS), Metastasis Free Survival (MFS), and Adverse Events (AE).
After a median follow up of 33.6 months, patients who received mFOLFIRINOX had a median DFS of 21.6 months compared with 12.8 months with Gemcitabine (HR=0.59; P<0.001) and the 3-year DFS was 39.7% with mFOLFIRINOX and 21.4% with Gemcitabine. The median OS was nearly 20 months longer with a mFOLFIRINOX regimen than with Gemcitabine (54.4 months versus 35 months). This represented a 34% reduction in the risk of death with mFOLFIRINOX (HR=0.66; P=0.003). The median MFS with mFOLFIRINOX regimen was 30.4 months versus 17.7 months with Gemcitabine (HR =0.59). Patients receiving mFOLFIRINOX experienced higher rates of grade 3 or 4 Adverse Events than with Gemcitabine for vomiting, diarrhea, fatigue, mucositis and sensory peripheral neuropathy. In the Gemcitabine group, the rate of grade 3/4 Adverse Events was higher for thrombocytopenia and febrile neutropenia.
It was concluded that adjuvant mFOLFIRINOX significantly improves Disease Free Survival, Metastasis Free Survival and Overall Survival, compared to Gemcitabine, after pancreatic cancer resection, in good Performance Status patients and should therefore be considered the new standard of care. It should be noted that patients with pancreatic cancer who undergo surgical resection, are fit enough to undergo this procedure and these patients would be the most likely candidates for mFOLFIRINOX. For those patients whose Performance Status is poor 12 weeks after surgery, and in those with clear contraindications to mFOLFIRINOX regimen, single agent Gemcitabine is an alternative treatment option. Unicancer GI PRODIGE 24/CCTG PA.6 trial: A multicenter international randomized phase III trial of adjuvant mFOLFIRINOX versus gemcitabine (gem) in patients with resected pancreatic ductal adenocarcinomas. Conroy T, Hammel P, Hebbar M, et al. J Clin Oncol 36, 2018 (suppl; abstr LBA4001)
FDA Approves BCL2 Inhibitor VENCLEXTA® for Chronic Lymphocytic Leukemia
SUMMARY: The FDA on June 8, 2018, granted regular approval to VENCLEXTA® (Venetoclax) for patients with Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL), with or without 17p deletion, who have received at least one prior therapy. The American Cancer Society estimates that for 2018, about 20,940 new cases of Chronic Lymphocytic Leukemia (CLL) will be diagnosed in the US and 4,510 patients will die of the disease. CLL accounts for about 25% of the new cases of leukemia and the average age at the time of diagnosis is around 71 years. B-cell CLL is the most common type of leukemia in adults.
The pro-survival (anti-apoptotic) protein BCL2 is over expressed by CLL cells and regulates clonal selection and cell survival. A new class of anticancer agents known as BH3-mimetic drugs mimic the activity of the physiologic antagonists of BCL2 and related proteins and promote apoptosis (programmed cell death). VENCLEXTA® is a second generation, oral, selective, small molecule inhibitor of BCL2 and restores the apoptotic processes in tumor cells. The FDA granted an accelerated approval to VENCLEXTA® in 2016, for the treatment of patients with CLL with 17p deletion, as detected by an FDA-approved test, who have received at least one prior therapy.
The present FDA approval was based on MURANO, which is an open-label, randomized, international, multicenter, phase III study which included 389 patients with Relapsed/Refractory CLL, who had received 1-3 prior lines of therapy, including at least one chemotherapy regimen. Patients were randomized 1:1 to receive a combination of either VR – VENCLEXTA® plus RITUXAN® (N=194) or BR – Bendamustine plus RITUXAN® (N=195). In the VR group, VENCLEXTA® tablets were given once daily with a weekly dose ramp-up schedule (20 mg for 1 week, followed by 1 week at each dose level of 50 mg, 100 mg, and 200 mg and then the recommended daily dose of 400 mg), over a period of 5 weeks, given along with Tumor Lysis Syndrome prophylaxis. Patients were treated with the 400 mg daily dosing for a maximum of 2 yrs or until disease progression. RITUXAN® (Rituximab) was given beginning week 6, and was administered at 375 mg/m2 on day 1, cycle 1, followed by 500 mg/m2 on day 1 of cycles 2 thru 6, of a 28 day cycle. In the BR group, Bendamustine was given at 70 mg/m2 on days 1 and 2 of each 28 day cycle for a total of 6 cycles along with RITUXAN®, using the same RITUXAN® dosing schedule as in the VR group. Patients were stratified based on del(17p) status and responsiveness to prior therapy. The median age was 65 years, 26% of the patients had del(17p) and 15% of the patients were refractory to Fludarabine. The Primary end point was Progression Free Survival (PFS) and Secondary end points included Overall Survival (OS), Overall Response Rate (ORR) and Complete Response (CR). The median follow up was 23.8 months.
It was noted that the median PFS was significantly superior in the VR group compared to BR and was not reached in the VR group and was 18.1 months in the BR group (HR=0.19; P<0.0001). The 2-year PFS rates were 84.9% versus 36.3%, respectively, favoring VENCLEXTA® (HR=0.17, P<0.0001). This meant an 83% reduction in the risk of disease progression or death in the VR group compared with the BR group. This PFS benefit was consistently seen in all subgroups assessed, including those with del(17p), p53 mutation and IgVH unmutated status. The 2-year PFS rate among patients with chromosome 17p deletion was 81.5% in the VR group versus 27.8% in the BR group (HR=0.13), and the 2-year PFS rate among those without chromosome 17p deletion was 85.9% versus 41.0% (HR=0.19). The Overall Response Rate, as assessed by the Independent Review Committee was 92.3% in the VR group, and 72.3% in the BR group (a difference of 20 percentage points). The rate of MRD (Minimal Residual Disease)-negativity based on peripheral blood samples, defined as less than 1 CLL cell in 10,000 leukocytes and attained at any time, was also higher with VR at 83.5% versus 23.1% with BR. Further, the MRD negativity was more durable in the VENCLEXTA® group. It has been noted that patients who are negative for MRD on the basis of peripheral blood sampling have better survival outcomes than patients who have a complete response and are positive for minimal residual disease. The high MRD clearance rate observed in the VR group exceed those previously attained with other regimens, in trials of Relapsed or Refractory CLL or SLL. The time to the next treatment for CLL was also longer in the VR group compared to BR group and at 2 years, 90% and 52.1%, respectively, had not received a next line of treatment for CLL (Hazard Ratio for receipt of next treatment or death= 0.19). Overall Survival evaluation is ongoing. Grade 3/4 neutropenia was higher in VR group but there was no increase in febrile neutropenia or Grade 3/4 infection.
It was concluded that VENCLEXTA® in combination with RITUXAN® resulted in a significant improvement in Progression Free Survival, Overall Response Rate, along with durable improvement in peripheral blood MRD negativity, when compared with Bendamustine and RITUXAN®, in patients with Relapsed/Refractory CLL. Venetoclax–Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. Seymour JF, Kipps TJ, Eichhorst B, et al. N Engl J Med 2018; 378:1107-1120
Late Breaking Abstract – ASCO 2018 Endocrine Therapy Alone is Adequate for Early Stage Breast Cancer Patients with Intermediate Risk Recurrence Score
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 266,120 new cases of invasive breast cancer will be diagnosed in 2018 and about 40,920 women will die of the disease. Approximately 50% of all breast cancers are Estrogen Receptor (ER) positive, HER2-negative, axillary node-negative tumors. Patients with early stage breast cancer often receive adjuvant chemotherapy. The Oncotype DX breast cancer assay, is a multigene genomic test that analyzes the activity of a group of 21 genes and is able to predict the risk of breast cancer recurrence and likelihood of benefit from systemic chemotherapy, following surgery, in women with early stage breast cancer. Chemotherapy recommendations for early stage, hormone receptor positive, HER negative, early stage breast cancer patients, are often made based on tumor size, grade, ImmunoHistoChemical (IHC) markers such as Ki-67, nodal status and Oncotype DX Recurrence Score (RS) assay.
Oncotype Dx assay categorizes patients on the basis of Recurrence Scores into Low risk (less than 18), Intermediate risk (18-30), and High risk (31 or more). It has been unclear whether patients in the Intermediate risk group benefited from the addition of chemotherapy to endocrine therapy. TAILORx was specifically designed to address this question and provide a very definitive answer. In this study, the Intermediate risk Recurrent Score (18-30) was changed to 11-25, to account for exclusion of higher-risk patients with HER2-positive disease and to minimize the potential for under treatment.
TAILORx ((Trial Assigning Individualized Options for Treatment) is a phase III, randomized, prospective, non-inferiority trial, and is the largest breast cancer treatment trial ever conducted, and the first precision medicine trial ever done, according to the authors. In this study, 10,273 women, 18-75 years of age, with hormone receptor-positive, HER2-negative, axillary node-negative breast cancer were enrolled. Patients had tumors 1.1-5.0 cm in size (or 0.6-1.0 cm and intermediate/high grade). Patients were divided into three groups based on their Recurrence Score. Women with a Low Recurrence Score of 0-10 received endocrine therapy alone and those with a High Recurrence Score of 26-100 received endocrine therapy in combination with standard adjuvant chemotherapy. Patient with Intermediate Recurrence Score of 11-25 (N=6711) were randomly assigned to receive endocrine therapy alone or endocrine therapy and adjuvant chemotherapy. The Primary endpoint was invasive Disease Free Survival, defined as recurrence of cancer in the breast, regional lymph nodes, and/or distant organs, a second primary cancer in the opposite breast or another organ, or death from any cause.
At a median follow-up of 7.5 years, the study met its Primary endpoint, and it was noted that that endocrine therapy alone was non-inferior to chemotherapy plus endocrine therapy, in patients with Intermediate Recurrence Score of 11-25. At 9 years, patients with Intermediate Recurrence Scores receiving endocrine therapy or chemotherapy in combination with endocrine therapy showed similar invasive Disease Free Survival rates (83.3% vs 84.3%), distant Recurrence Free Interval (94.5% vs 95.0%), Recurrence Free Interval (92.2% vs 92.9%) and Overall Survival (93.9% vs 93.8%) respectively. These findings suggested that there was no benefit from adding chemotherapy to endocrine therapy, for this patient group.
The authors also conducted an exploratory analysis of patients in the Intermediate Risk group to determine which patients would benefit from added chemotherapy. They noted that there was no significant interaction between menopause, tumor size or grade, with Recurrence Score. There was however an interaction between age and Recurrence Score. In women 50 years or younger with a Recurrence Score of 16-20, there were 2% fewer distant recurrences, and in those with a recurrence score of 21-25, there were 7% fewer distant recurrences with the addition of chemotherapy, suggesting that younger women with a Recurrence Score of 16-25 had some benefit with the addition of chemotherapy to endocrine therapy.
It was concluded that women older than 50 years with hormone receptor-positive, HER2-negative, node-negative breast cancer and a Recurrence Score of 0-25, as well as women 50 years or younger with hormone receptor-positive, HER2-negative, node-negative breast cancer and a Recurrence Score of 0-15, could be spared from chemotherapy, based on this study. This study showed that chemotherapy could be avoided in about 70% of these patients, by allowing this test to tailor treatment. Further, this prospective study reflects outcomes with current modern chemotherapy and endocrine therapy regimens. The authors recommended that any patient 75 years or younger with early-stage breast cancer should therefore be offered Oncotype DX assay test, for guidance regarding chemotherapy recommendations after surgery. TAILORx: phase III trial of chemoendocrine therapy versus endocrine therapy alone in hormone receptor-positive, HER2-negative, node-negative breast cancer and an intermediate prognosis 21-gene recurrence score. Sparano JA, Gray RJ, Wood WC, et al. J Clin Oncol. 2018;36(suppl; abstr LBA1).
MRI Targeted Biopsy Superior to Standard TRUS Guided Biopsy for Prostate Cancer Diagnosis
TransRectal UltraSound (TRUS) guided biopsy has been the standard of care for diagnosing prostate cancer in men with a clinical suspicion of prostate cancer. TRUS guided biopsy is a blind biopsy of the lateral and posterior peripheral zone of the prostate using a template, and 10 to 12 cores of prostate tissue is obtained. Even though this may result in a higher rate of prostate cancer detection, many detected are low grade tumors that do not benefit from treatment. The major limitation of this biopsy procedure is the risk of under-sampling a more significant tumor that is located in a region of the prostate not usually targeted with a template.
Multiparametric MRI (mp-MRI) combines anatomic imaging in the form of T2-weighted imaging, with functional imaging, and is being used to detect or rule out cancer in men who have persistent concern for prostate cancer. It can be used as a triage test to avoid a biopsy if the results were negative, and if positive could be used for targeting abnormal areas in the prostate during biopsy. In the PRECISION study, mp-MRI was superior to standard TRUS guided biopsy, and was able to identify a high proportion of men who would benefit from treatment, and minimizes the identification of men with clinically insignificant cancer, thereby preventing overtreatment. Utilizing mp-MRI, more than 25% of the participants in this study were able to avoid a biopsy.