
The FDA on September 14, 2017 granted accelerated approval to ALIQOPA®, for the treatment of adult patients with relapsed Follicular Lymphoma, who have received at least two prior systemic therapies. ALIQOPA® is a product of Bayer HealthCare Pharmaceuticals Inc.
Author: RR
JEVTANA® (Cabazitaxel)
The FDA on September 14, 2017 approved a lower dose of JEVTANA® (20 mg/m2 every 3 weeks) in combination with Prednisone for the treatment of patients with metastatic Castration-Resistant Prostate Cancer, previously treated with a Docetaxel-containing treatment regimen. JEVTANA® (25 mg/m2 every 3 weeks) was approved for this indication in 2010. JEVTANA® is a product of Sanofi-Aventis.
BESPONSA® (Inotuzumab ozogamicin)
The FDA on August 17, 2017 approved BESPONSA® for the treatment of adults with relapsed or refractory B-cell precursor Acute Lymphoblastic Leukemia (ALL). BESPONSA® is a product of Wyeth Pharmaceuticals Inc., a subsidiary of Pfizer Inc.
LYNPARZA® (Olaparib)
The FDA on August 17, 2017 granted regular approval to LYNPARZA® tablets for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a complete or partial response to platinum-based chemotherapy. LYNPARZA® is a product of AstraZeneca.
MYLOTARG® (Gemtuzumab ozogamicin)
The FDA on September 1, 2017 approved MYLOTARG® for the treatment of newly-diagnosed CD33-positive Acute Myeloid Leukemia (AML) in adults and for treatment of relapsed or refractory CD33-positive AML in adults and in pediatric patients 2 years and older. MYLOTARG® may be used in combination with Daunorubicin and Cytarabine for adults with newly diagnosed AML, or as a stand-alone treatment for certain adult and pediatric patients. MYLOTARG® is a product of Pfizer Inc.
FDA Approves ALIQOPA®, a PI3K Inhibitor, for Follicular Lymphoma
SUMMARY: The FDA on September 14, 2017, granted accelerated approval to ALIQOPA® (Copanlisib) for the treatment of adult patients with relapsed Follicular Lymphoma, who have received at least two prior systemic therapies. The American Cancer Society estimates that in 2017, about 72,240 people will be diagnosed with Non Hodgkin Lymphoma (NHL) in the United States and about 20,140 individuals will die of this disease. Indolent NHLs are mature B cell lymphoproliferative disorders and include Follicular Lymphoma, Nodal Marginal Zone Lymphoma (NMZL), Extranodal Marginal Zone Lymphoma (ENMZL) of Mucosa-Associated Lymphoid Tissue (MALT), Splenic Marginal Zone Lymphoma (SMZL), LymphoPlasmacytic Lymphoma (LPL) and Small Lymphocytic Lymphoma (SLL). Follicular Lymphoma is the most indolent form and second most common form of all NHLs and they are a heterogeneous group of lymphoproliferative malignancies. Approximately 20% of all NHLs are Follicular Lymphomas. Advanced stage indolent NHL is not curable and as such, prolonging Progression Free Survival (PFS) and Overall Survival (OS), while maintaining Quality of Life, have been the goals of treatment intervention. Asymptomatic patients with indolent NHL are generally considered candidates for “watch and wait” approach whereas those with B symptoms (fever, night sweats, and weight loss), painful lymphadenopathy/splenomegaly, organ compromise and cytopenias are generally considered candidates for therapy.. Follicular Lymphoma International Prognostic Index (FLIPI) is of prognostic value and is used to help with treatment choices. The Ann Arbor classification divides FL into four stages. Patients with stages I and II have localized disease and those with stages III and IV have advanced disease. The World Health Organization (WHO) further classified FL based on histology into low grade (grades 1 and 2) and high grade (grade 3a) FLs. Grade 3b FL which demonstrates diffuse areas of involvement is designated as Diffuse Large B-cell Lymphoma (DLBCL) and is treated as such. Patients with advanced stage symptomatic Follicular Lymphoma are often treated with induction chemoimmunotherapy followed by maintenance RITUXAN® (Rituximab). This can result in a median PFS of 6-8 yrs and a median Overall Survival of 12-15 yrs. However, approximately 30% of the patients will relapse in 3 years and treatment options are limited for patients with relapses, after multiple treatments.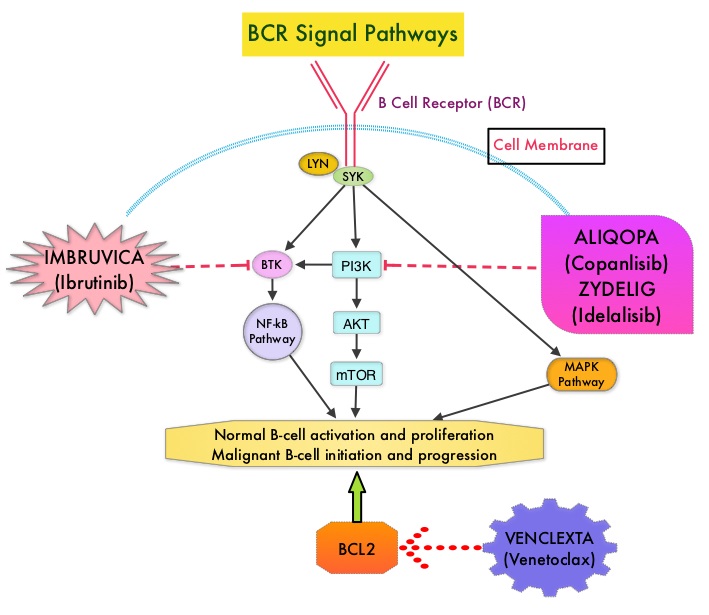
ALIQOPA® is a pan-class 1, PI3K inhibitor with inhibitory activity predominantly against PI3K-α and PI3K-δ Isoforms expressed in malignant B cells. The alpha isoform is broadly expressed and involved in insulin signaling and angiogenesis, as well as resistance mechanisms to lymphoma whereas the delta isoform is expressed by leukocytes and is involved in B-cell signaling, development, and survival. ALIQOPA® has been shown to induce tumor cell death by apoptosis and inhibition of proliferation of primary malignant B cell lines. ALIQOPA® also inhibits several key cell-signaling pathways, including B-cell receptor (BCR) signaling, CXCR12 (C-X-C chemokine receptor 12) mediated chemotaxis of malignant B cells, and NFÏ°B (Nuclear Factor Kappa B) signaling in lymphoma cell lines.
The approval of ALIQOPA® was based on data from the CHRONOS-1 trial, which is an open-label, single arm, multicenter, phase II study of patients with relapsed, Indolent or aggressive Non Hodgkin Lymphomas. This trial included patients with Follicular lymphoma (Grades 1-3a), Marginal Zone Lymphoma, Small Lymphocytic Lymphoma, and LymphoPlasmacytic Lymphoma /Waldenstrom Macroglobulinemia . Eligible patients had relapsed or refractory disease and had received at least two prior systemic therapies. The efficacy data leading to the FDA approval included 104 patients with Follicular B-cell Non Hodgkin Lymphoma. In this trial, patients received 0.8 mg/kg or 60 mg of ALIQOPA® by IV infusion on days 1, 8, and 15 of a 28-day treatment cycle, until disease progression or development of unacceptable toxicity. The median patient age was 63 yrs and all study patients had prior exposure to RITUXAN® and one or more alkylating agents, and 60% of the patients had disease that was refractory to the last regimen received. The Primary endpoint was Objective Response Rate after a minimum of 16 weeks of treatment. Secondary endpoints included Progression Free Survival, Duration of Response, Overall Survival, safety, and Quality of Life.
The Objective Response Rate was 58.7%, with an estimated median response duration of 12.2 months. The Complete Response rate was 14.4% and partial response rate was 44.3% and 33.7% has stable disease. The most common adverse reactions included nausea, hyperglycemia, diarrhea, fatigue, hypertension, cytopenias and lower respiratory tract infections. According to the authors, safety was manageable compared with the other PI3K inhibitor approved by the FDA, ZYDELIG® (Idelalisib), which targets only the δ-isoform and has warnings against fatal or severe colitis, intestinal perforation, hepatotoxicity, and pneumonitis The safety advantage with ALIQOPA® may be due to the dose scheduling or the IV mode of delivery.
It was concluded that ALIQOPA® has significant activity in patients with relapsed/refractory indolent B-cell lymphoma, and the safety was manageable, compared with other PI3K inhibitors. Two phase III trials are underway, using ALIQOPA® in combination with RITUXAN®. Dreyling M, Santoro A, Mollica L, et al. Copanlisib in patients with relapsed or refractory indolent B-cell lymphoma: Primary results of the pivotal CHRONOS-1 study. Presented at: 2017 AACR Annual Meeting; April 1-5, 2017; Washington, DC. Abstract CT149.
FDA Approves KEYTRUDA® for Advanced Gastric Cancer
SUMMARY: The FDA on September 22, 2017 granted accelerated approval to KEYTRUDA® (Pembrolizumab), for patients with recurrent locally advanced or metastatic, Gastric or GastroEsophageal junction adenocarcinoma, whose tumors express PD-L1, as determined by an FDA-approved test. Patients must have had disease progression on or after two or more prior systemic therapies, including Fluoropyrimidine and platinum-containing chemotherapy and, if appropriate, HER2/neu-targeted therapy. Cancers of the esophagus and stomach are among the most prevalent malignancies and are a major cause of cancer-related mortality. It is estimated that in 2016 GastroEsophageal Adenocarcinoma (GEA) accounted for 43,280 new cases in the United States. The incidence of these tumors has been on the rise in the past decade. Majority of the patients with GEA have advanced disease at the time of initial presentation and have limited therapeutic options with little or no chance for cure. Patients with localized disease (stage II and stage III) are often treated with multimodality therapy and 40% of the patients may survive for 5 years or more.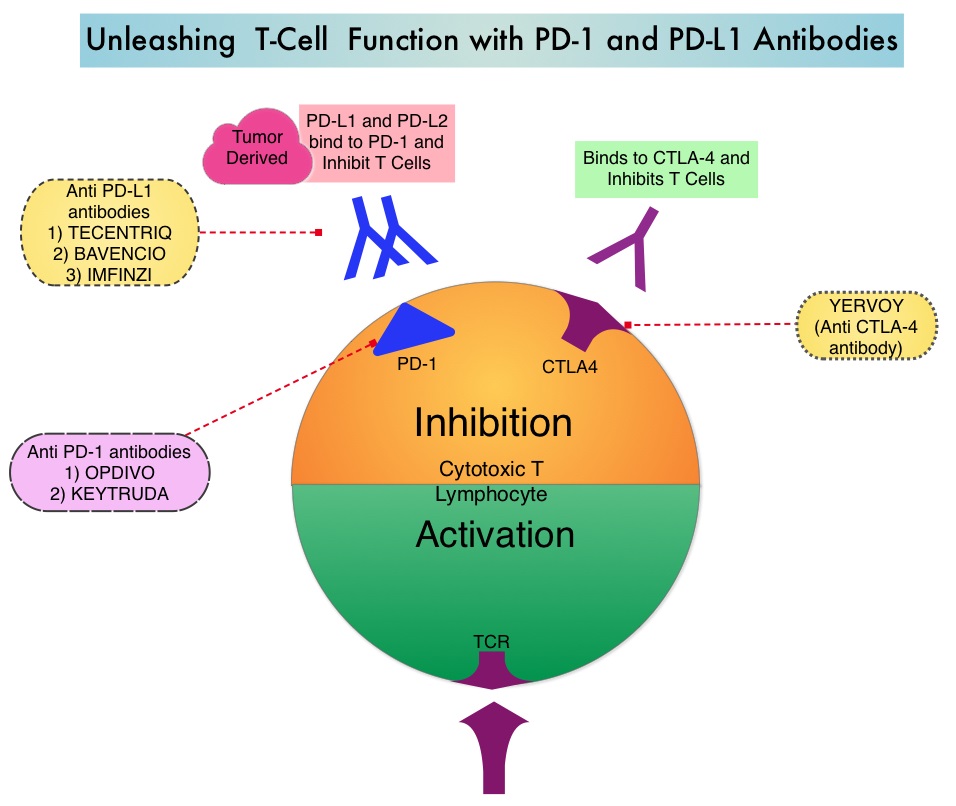
KEYTRUDA® is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the tumor-specific effector T cells. In previously reported Phase I study, KEYTRUDA® showed promising antitumor activity with manageable safety, in patients with previously treated advanced gastric cancer.
Based on these findings, the authors conducted a global, multicohort, Phase II study (KEYNOTE 059), in which patients with advanced gastric or gastroesophageal junction cancer received KEYTRUDA® 200 mg every 3 weeks for up to 2 years or until disease progression or unacceptable toxicity. Cohort 1 in this study enrolled 259 patients of whom 57% (N=148) had PD-L1 positive tumors and these tumors were either MicroSatellite Stable (MSS), or had undetermined MicroSatellite Instability (MSI) or MisMatch Repair (MMR) status. PD-L1 expression was evaluated by the PD-L1 IHC (22C3 antibody) and PD-L1 positivity was based on a Combined Positive Score (CPS) of 1 or more. CPS is determined by the number of PD-L1 staining cells (tumor cells, lymphocytes, macrophages) divided by total number of tumor cells evaluated, multiplied by 100. The median age was 62 years, 76% were men and over 50% of the patients received KEYTRUDA® as third line treatment or beyond. The median duration of follow up was 5.4 months. Primary end points included Objective Response Rate (ORR), safety, and tolerability.
In the group of patients with PD-L1 positive tumors, the ORR was 15.5% with 2% Complete Responses and 13.5% Partial Responses. Among the responding patients, the response duration ranged from 3-19 months, with 58% of the responding patients having response durations of 6 months or longer and 26% of the responding patients having response durations of 12 months or longer. In this cohort of 259 patients, 3% had tumors that were determined to be MSI-High. The ORR in this group was 57%, with response duration ranging from 5-14 months. In the PD-L1 negative patients however, the ORR was only 5.5%.
It was concluded that KEYTRUDA® demonstrated promising antitumor activity and durable responses in patients with advanced Gastric/GastroEsophageal Junction cancer, who had progressed on more than 2 lines of therapy, with higher Objective Response Rates noted in patients with PD-L1-positive tumors. KEYNOTE-059 cohort 1: Efficacy and safety of pembrolizumab (pembro) monotherapy in patients with previously treated advanced gastric cancer. Fuchs CS, Doi T, Jang RW-J, et al. J Clin Oncol. 2017;35 (suppl; abstr 4003).
FDA Approves NERLYNX® for Adjuvant Treatment of HER2 Positive Breast Cancer
The FDA on July 17, 2017 approved NERLYNX® (Neratinib) for the extended adjuvant treatment of adult patients with early stage HER2-overexpressed/amplified breast cancer, to follow adjuvant Trastuzumab (HERCEPTIN®)-based therapy. NERLYNX® is a potent, irreversible, oral Tyrosine Kinase Inhibitor, of HER1, HER2 and HER4 (pan-HER inhibitor). NERLYNX® is the first TKI approved by the FDA, shown to reduce the risk for disease recurrence, in patients with early stage HER2-positive breast cancer and demonstrated significantly improved 2-year invasive Disease Free Survival.
FDA Approves First Biosimilar for Cancer Treatment
SUMMARY: The FDA on Sept. 14, 2017 approved MVASI® (Bevacizumab-awwb) as a Biosimilar to AVASTIN® (Bevacizumab). Bevacizumab is a recombinant immunoglobulin G1 (IgG1) monoclonal antibody (mAb) that binds to Vascular Endothelial Growth Factor (VEGF) and inhibits the interaction of VEGF with its receptors, VEGF receptor-1 and VEGF receptor-2. This in turn inhibits establishment of new blood vessels that are essential for the maintenance and growth of solid tumors. MVASI® is the first Biosimilar approved in the U.S. for the treatment of cancer. A Biosimilar product is a biological product that is approved based on its high similarity to an already approved biological product (also known as reference product). Biological products are made from living organisms including humans, animals and microorganisms such as bacteria or yeast, and are manufactured through biotechnology, derived from natural sources or produced synthetically. Biological products have larger molecules with a complex structure, than conventional drugs (also known as small molecule drugs).
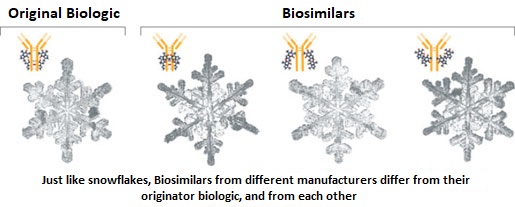 Unlike biological products, conventional drugs are made of pure chemical substances and their structures can be identified. A generic drug is a copy of brand name drug and has the same active ingredient and is the same as brand name drug in dosage form, safety and strength, route of administration, quality, performance characteristics and intended use. Therefore, brand name and the generic drugs are bioequivalent. The Affordable Care Act in 2010 created an abbreviated licensure pathway for biological products that are demonstrated to be “Biosimilar” to, or “interchangeable” with an FDA approved biological product (reference product). The Biosimilar must show that it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. A Biosimilar product can only be approved by the FDA if it has the same mechanism of action, route of administration, dosage form and strength as the reference product, and only for the indications and conditions of use that have been approved for the reference product. Biosimilars are not as easy to manufacture as generics (copies of brand name drugs), because of the complexity of the structure of the biologic product and the process used to make a biologic product. The facilities where Biosimilars are manufactured must also meet the FDA standards.
Unlike biological products, conventional drugs are made of pure chemical substances and their structures can be identified. A generic drug is a copy of brand name drug and has the same active ingredient and is the same as brand name drug in dosage form, safety and strength, route of administration, quality, performance characteristics and intended use. Therefore, brand name and the generic drugs are bioequivalent. The Affordable Care Act in 2010 created an abbreviated licensure pathway for biological products that are demonstrated to be “Biosimilar” to, or “interchangeable” with an FDA approved biological product (reference product). The Biosimilar must show that it has no clinically meaningful differences in terms of safety and effectiveness from the reference product. A Biosimilar product can only be approved by the FDA if it has the same mechanism of action, route of administration, dosage form and strength as the reference product, and only for the indications and conditions of use that have been approved for the reference product. Biosimilars are not as easy to manufacture as generics (copies of brand name drugs), because of the complexity of the structure of the biologic product and the process used to make a biologic product. The facilities where Biosimilars are manufactured must also meet the FDA standards.
MVASI® is approved for the treatment of patients with the following cancers:
• Metastatic Colorectal cancer, in combination with intravenous 5-Fluorouracil-based chemotherapy for first or second line treatment. MVASI® is not indicated for the adjuvant treatment of surgically resected Colorectal cancer.
• Metastatic Colorectal cancer, in combination with Fluoropyrimidine-Irinotecan or Fluoropyrmidine-Oxaliplatin-based chemotherapy for the second line treatment of patients who have progressed on a first-line Bevacizumab containing regimen.
• Non-squamous Non Small Cell Lung Cancer, in combination with Carboplatin and Paclitaxel for first line treatment of unresectable, locally advanced, recurrent or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in Objective Response Rate. No data is available demonstrating improvement in disease-related symptoms or survival with Bevacizumab.
• Metastatic Renal cell carcinoma, in combination with Interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic disease, in combination with Paclitaxel and Cisplatin or Paclitaxel and Topotecan.
The approval of MVASI® was based on two studies. In the first study, PharmacoKinetics (PK) of biosimilar MVASI® was compared with Bevacizumab, following a single infusion of 3 mg/kg. It was concluded that the PK data was similar between the Biosimilar, MVASI® and Bevacizumab. The second study is a randomized, double-blind, phase III trial, that evaluated the efficacy and safety of MVASI®, compared with Bevacizumab, in patients with non-squamous Non Small Cell Lung Cancer (NSCLC). Patients with non-squamous NSCLC, on first line chemotherapy with Carboplatin and TAXOL® (Paclitaxel), were randomized in a 1:1 ratio to receive either MVASI® (N=328) or Bevacizumab 15 mg/kg (N=314), as an IV infusion, every 3 weeks, for 6 cycles. The Objective Response Rate (ORR) was similar between the two treatment groups (39.0% for MVASI® and 41.7% for Bevacizumab) and these results were not statistically different. The Duration of Response was similar. Adverse events were comparable in the two treatment groups. This study demonstrated that MVASI® was clinically similar to Bevacizumab.
The FDA concluded that the approval of MVASI® was based on comparisons of extensive structural and functional product characterization, animal data, human PharmacoKinetic and pharmacodynamic data, clinical immunogenicity, between MVASI® and AVASTIN® (Bevacizumab), and it was noted that MVASI® is highly similar to AVASTIN® and that there are no clinically meaningful differences between the two products. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. Thatcher N, Thomas M, Paz-Ares L, et al. DOI: 10.1200/JCO.2016.34.15_suppl.9095 Journal of Clinical Oncology 34, no. 15_suppl (May 2016) 9095-9095.
E-Cigarettes Can Increase the Risk of Bladder Cancer
SUMMARY: The American Cancer Society estimates that tobacco use is responsible for nearly 1 in 5 deaths in the United States and accounts for at least 30% of all cancer deaths. Smokeless tobacco products are a major source of cancer causing nitrosamines, and increase the risk of developing cancer of the oropharynx, esophagus, and pancreas. Cigarette smoke contains more than 7,000 chemicals, many of which are toxic and some linked to cancer. E-cigarettes or Electronic Nicotine Delivery Systems (ENDS) were first developed in China and introduced to the U.S. market in 2007. When a smoker inhales through the mouth piece of an e-cigarette, the air flow triggers a sensor that switches on a small lithium battery powered heater, which in turn vaporizes liquid nicotine along with PolyEthylene Glycol (PEG) present in a small cartridge. The PEG vapor looks like smoke. The potent liquid form of nicotine extracted from tobacco is tinctured with fragrant flavors such as chocolate, cherry and bubble gum, coloring substances, as well other chemicals and these e-liquids are powerful neurotoxins.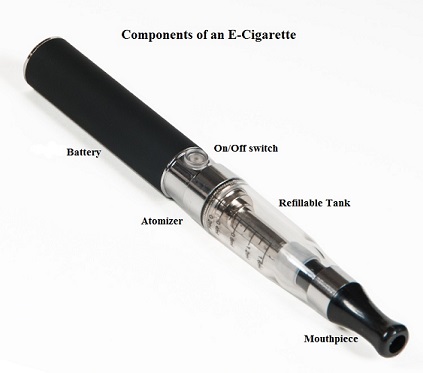
With the rapid growth of the e-cigarette industry and the evidence of potential dangers and risk to public health, particularly children, experts from the world's leading lung organizations were compelled to release a position statement on electronic cigarettes, specifically focusing on their potential adverse effects on human health and calling on government organizations to ban or restrict the use of e-cigarettes, until their impact on health is better understood. According to the National Youth Tobacco Survey, the use of e-cigarettes has tripled from 2013 to 2014 among middle school and high school students. Epidemiological data have shown that nicotine use is a gateway to the use of cocaine and marijuana and subsequent lifelong addiction. E-cigarettes and other Electronic Nicotine Delivery Systems (ENDS), unlike combustible cigarettes and many other tobacco products are not currently regulated by the U.S. Food and Drug Administration.
Smoking more than one pack of cigarettes a day has been associated with an increased risk of mortality among patients with bladder cancer. E-cigarettes have been advertised and promoted as a safer way of delivering the stimulating effects of tobacco smoke, without the harmful health risks. Contrary to this claim, over 90% of inhaled nicotine is excreted to urine. Nicotine can be nitrosatized in urothelial cells and then further metabolized into carcinogenic nitrosamines and formaldehyde, which in turn can induce DNA damage in the bladder mucosa. Additionally, these metabolites can also block DNA repair, increasing cancer risk.
The authors in this study compared the urine of e-cigarette users to that of nonsmokers, for known bladder carcinogens. There are five known bladder carcinogens that are either present in traditional cigarettes or common solvents believed to be used in some e-cigarette formulations, and they include (benz(a)anthracene, benzo(a)pyrene, 1-hydroxypyrene, o-toluidine and 2-naphthylamine. The limit of detection of these carcinogens in this study was 10-100 ng/ml. Urine samples were collected from 13 e-cigarette users and 10 non-smoking, non e-cigarette using controls. The mean age was 39 years and participants were predominantly male and had abstained completely from traditional cigarettes, for at least 6 months prior to specimen collection.
It was noted that the urine from 92% of e-cigarette users tested positive for two of the five carcinogenic compounds whereas none of the controls tested positive for these carcinogens. Interestingly, all study participants considered e-cigarettes were safe, when interviewed by the study investigators.
It was concluded that in this important pilot study, majority of the e-cigarette users had carcinogenic metabolites in the urine, which can contribute to the development of bladder cancer. This study underscores the importance both traditional and e-cigarette smoking cessation, for bladder cancer prevention. MP88-14 EVALUATION OF E-CIGARETTES USERS URINE FOR KNOWN BLADDER CARCINOGENS. Fuller T, Acharya A, Bhaskar G, et al. DOI: http://dx.doi.org/10.1016/j.juro.2017.02.2739