
The FDA on December 3, 2014 granted accelerated approval for BLINCYTO® for the treatment of Philadelphia chromosome-negative relapsed or refractory B-cell precursor Acute Lymphoblastic Leukemia (R/R ALL). BLINCYTO® is a product of Amgen Inc.
Author: RR
AVASTIN® (Bevacizumab)
The FDA on November 14, 2014 approved AVASTIN® in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan for the treatment of patients with platinum-resistant, recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer. AVASTIN® is a product of Genentech, Inc.
Sentinel Lymph Node Biopsy for Patients with Early-Stage Breast Cancer American Society of Clinical Oncology Clinical Practice Guideline Update
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately, 233,000 new cases of invasive breast cancer will be diagnosed in 2014 and 40,000 women will die of the disease. Surgical resection of the axillary lymph nodes in addition to potentially removing cancer that may have spread, also facilitates staging of breast cancer. 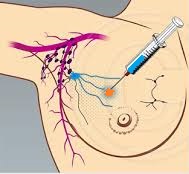 The sentinel node is the first lymph node(s) to which cancer cells are most likely to metastasize from a primary tumor. With the introduction of intraoperative lymphatic mapping in the 1990s, Sentinel Lymph Node Biopsy (SLNB) has gained general acceptance and is the preferred procedure in appropriate circumstances. Unlike Axillary Lymph Node Dissection (ALND), SLNB is associated with a lower incidence of Lymphedema, seroma at the surgery site, paresthesias and restriction of joint movement. Nine randomized clinical trials have not shown any difference in mortality among patients who underwent ALND or SLNB for either lymph node metastases or negative sentinel lymph nodes, validating Sentinel Lymph Node Biopsy (SLNB). The American Society of Clinical Oncology (ASCO) first published guidelines on the use of SLNB for patients with early stage breast cancer in 2005, based on one randomized clinical trial. Since then, additional information from 9 randomized clinical trials and13 cohort studies pertinent to SLNB and ALND has resulted in this ASCO Clinical Practice Guideline Update.
The sentinel node is the first lymph node(s) to which cancer cells are most likely to metastasize from a primary tumor. With the introduction of intraoperative lymphatic mapping in the 1990s, Sentinel Lymph Node Biopsy (SLNB) has gained general acceptance and is the preferred procedure in appropriate circumstances. Unlike Axillary Lymph Node Dissection (ALND), SLNB is associated with a lower incidence of Lymphedema, seroma at the surgery site, paresthesias and restriction of joint movement. Nine randomized clinical trials have not shown any difference in mortality among patients who underwent ALND or SLNB for either lymph node metastases or negative sentinel lymph nodes, validating Sentinel Lymph Node Biopsy (SLNB). The American Society of Clinical Oncology (ASCO) first published guidelines on the use of SLNB for patients with early stage breast cancer in 2005, based on one randomized clinical trial. Since then, additional information from 9 randomized clinical trials and13 cohort studies pertinent to SLNB and ALND has resulted in this ASCO Clinical Practice Guideline Update.
The following recommendations were made by the American Society of Clinical Oncology panel of experts:
1) Women without sentinel lymph node (SLN) metastases should not undergo Axillary Lymph Node Dissection (ALND).
2) Women with one to two metastatic SLNs planning to undergo breast conserving surgery with whole breast radiotherapy should not undergo ALND (in most cases).
3) Women with SLN metastases who will undergo mastectomy should be offered ALND.
4) Women with operable breast cancer and multicentric tumors, those with ductal carcinoma in situ (DCIS) who will undergo mastectomy, those who previously underwent breast and/or axillary surgery and those who received preoperative/neoadjuvant systemic therapy, may be offered SLNB.
5) Women who have large or locally advanced invasive breast cancer (tumor size T3/T4), inflammatory breast cancer, or DCIS (when breast-conserving surgery is planned) or are pregnant, should not undergo SLNB.
Lyman GH, Temin S, Edge SB, et al. J Clin Oncol 2014;32:1365-1383
Hepatitis B Reactivation in Patients with Previous Hepatitis B Virus Exposure Undergoing Rituximab-Containing Chemotherapy for Lymphoma A Prospective Study
SUMMARY:The Centers for Disease Control and Prevention (CDC) estimates that there are 800,000 -1.4 million individuals with Chronic Hepatitis B infection in the United States. Reactivation of HBV is a major concern in cancer patients who may be on chemotherapy or other immunosuppressive therapies, with the incidence of HBV reactivation ranging from 40%-60% in those who are positive for Hepatitis B surface antigen (HBsAg). HBV reactivation is preventable with prophylactic antiviral therapy, failing which it can result in delays in cancer treatment as well as potentially fatal outcomes.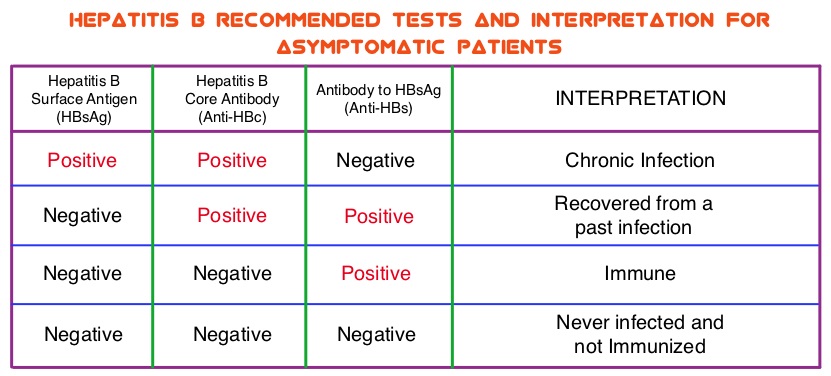 The CDC updated their recommendations in 2008 and recommended HBV screening for patients receiving cytotoxic chemotherapy or immunotherapy. The American Society of Clinical Oncology in 2010 rendered a Provisional Clinical Opinion (PCO) suggesting that there was insufficient evidence to recommend routine screening for HBV in cancer patients, but screening may be considered for patient populations at high risk or for those who are to receive highly immunosuppressive therapies including anti-CD20 monoclonal antibody therapy such as RITUXAN® (Rituximab). According to the International recommendations, HBV reactivation is defined as the detection of serum HBV DNA of 10 IU/mL or more, by a real-time polymerase chain reaction–based assay. Because of the ambiguity regarding HBV reactivation in lymphoma patients receiving immunosuppressive therapy, the authors conducted a prospective trial to determine the frequency and factors predictive of HBV reactivation in HBsAg-negative, anti-HBc–positive patients treated with RITUXAN® based chemotherapy regimens. In this observational study, 260 patients with hematologic malignancies who were HBsAg-negative, anti-HBc–positive, with undetectable serum HBV DNA (< 10 IU/mL) and treated with RITUXAN® containing chemotherapy, were prospectively monitored every 4 weeks for up to 2 years. Patients were started on BARACLUDE® (Entecavir), when HBV reactivation (serum HBV DNA of 10 IU/mL or more), was documented.
The CDC updated their recommendations in 2008 and recommended HBV screening for patients receiving cytotoxic chemotherapy or immunotherapy. The American Society of Clinical Oncology in 2010 rendered a Provisional Clinical Opinion (PCO) suggesting that there was insufficient evidence to recommend routine screening for HBV in cancer patients, but screening may be considered for patient populations at high risk or for those who are to receive highly immunosuppressive therapies including anti-CD20 monoclonal antibody therapy such as RITUXAN® (Rituximab). According to the International recommendations, HBV reactivation is defined as the detection of serum HBV DNA of 10 IU/mL or more, by a real-time polymerase chain reaction–based assay. Because of the ambiguity regarding HBV reactivation in lymphoma patients receiving immunosuppressive therapy, the authors conducted a prospective trial to determine the frequency and factors predictive of HBV reactivation in HBsAg-negative, anti-HBc–positive patients treated with RITUXAN® based chemotherapy regimens. In this observational study, 260 patients with hematologic malignancies who were HBsAg-negative, anti-HBc–positive, with undetectable serum HBV DNA (< 10 IU/mL) and treated with RITUXAN® containing chemotherapy, were prospectively monitored every 4 weeks for up to 2 years. Patients were started on BARACLUDE® (Entecavir), when HBV reactivation (serum HBV DNA of 10 IU/mL or more), was documented.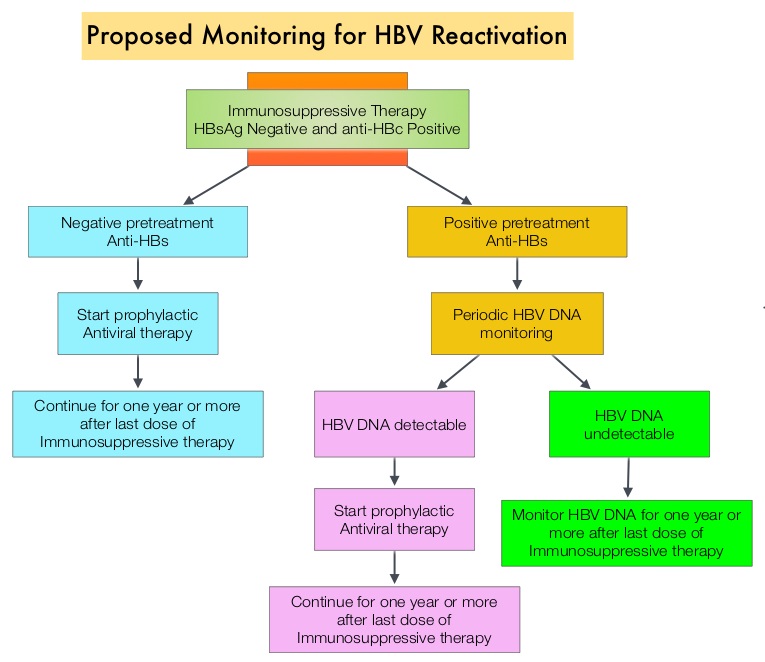 The cumulative rate of HBV reactivation over the 2 year observation period was high at 41.5%. The HBV reactivation occurred at a median of 23 weeks after RITUXAN® treatment and the median HBV DNA level at reactivation was 43 IU/mL. Undetectable antibody level to HBsAg (anti-HBs; < 10 mIU/mL) at baseline, prior to treatment with RITUXAN®, was the only significant risk factor that was strongly associated with HBV reactivation (P=0.009). Patients with negative baseline antibody level to HBsAg (anti-HBs) had a significantly higher 2-year cumulative rate of HBV reactivation, compared with those who had positive baseline antibody level to HBsAg (68.3% vs 34.4%; P=0.012). All patients had normal ALT when HBV reactivation occurred and except for one patient, were HBsAg negative. More importantly, all patients with HBV reactivation were successfully treated with BARACLUDE®. The authors concluded that HBsAg-negative, anti-HBc–positive lymphoma patients, receiving RITUXAN® based chemotherapy regimens experience a high rate of HBV reactivation, with this rate even significantly higher in patients with negative baseline antibody level to HBsAg. Periodic monitoring for HBV reactivation can enable early detection and intervention,thereby avoiding HBV related morbidities and mortality. Seto W, Chan T, Hwang Y, et al. JCO 2014;32:3736-3743
The cumulative rate of HBV reactivation over the 2 year observation period was high at 41.5%. The HBV reactivation occurred at a median of 23 weeks after RITUXAN® treatment and the median HBV DNA level at reactivation was 43 IU/mL. Undetectable antibody level to HBsAg (anti-HBs; < 10 mIU/mL) at baseline, prior to treatment with RITUXAN®, was the only significant risk factor that was strongly associated with HBV reactivation (P=0.009). Patients with negative baseline antibody level to HBsAg (anti-HBs) had a significantly higher 2-year cumulative rate of HBV reactivation, compared with those who had positive baseline antibody level to HBsAg (68.3% vs 34.4%; P=0.012). All patients had normal ALT when HBV reactivation occurred and except for one patient, were HBsAg negative. More importantly, all patients with HBV reactivation were successfully treated with BARACLUDE®. The authors concluded that HBsAg-negative, anti-HBc–positive lymphoma patients, receiving RITUXAN® based chemotherapy regimens experience a high rate of HBV reactivation, with this rate even significantly higher in patients with negative baseline antibody level to HBsAg. Periodic monitoring for HBV reactivation can enable early detection and intervention,thereby avoiding HBV related morbidities and mortality. Seto W, Chan T, Hwang Y, et al. JCO 2014;32:3736-3743
Randomized phase 3 study of rituximab, cyclophosphamide, doxorubicin, and prednisone plus vincristine (R-CHOP) or bortezomib (VR-CAP) in newly diagnosed mantle cell lymphoma (MCL) patients (pts) ineligible for bone marrow transplantation (BMT)
SUMMARY: The U.S. Food and Drug Administration (FDA) on October 9, 2014, approved VELCADE® (Bortezomib), a proteasome inhibitor, as combination regimen, for use in previously untreated patients with Mantle Cell Lymphoma (MCL). Non-Hodgkin Lymphoma (NHL) is one of the most common cancers in the United States and the American Cancer Society estimates that in 2014, about 70,800 people will be diagnosed with NHL in the US and close to 19,000 people will die of the disease. Mantle Cell Lymphomas constitute approximately 5% of all Non Hodgkin lymphomas and have a high relapse rate following dose-intensive therapies. VELCADE® was initially approved by the FDA in 2006 for the treatment of relapsed or refractory Mantle Cell Lymphoma and has a response rate of 30%. This latest approval was based on the results of an international, randomized, open-label phase III trial in which 487 patients with stage II to IV MCL, who were ineligible or not considered for Bone Marrow Transplantation, received VR-CAP (N = 243) or R-CHOP (N = 244). VR- CAP is essentially R-CHOP with the Vincristine replaced by VELCADE®. So, VR-CAP regimen consisted of VELCADE® administered IV at 1.3 mg/m2 on days 1, 4, 8, and 11, RITUXAN® (Rituximab) 375 mg/m2 IV given on day 1, Cyclophosphamide 750 mg/m2 IV on day 1, Doxorubicin 50 mg/m2 IV on day 1 and Prednisone at 100 mg/m2 PO on days 1 to 5 of a 21 day cycle for 6-8 cycles. R-CHOP regimen was exactly similar except that Vincristine 1.4 mg/m2 (max 2 mg) IV was administered on day 1 of each cycle instead of VELCADE®. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Time To Progression (TTP), Time To Next Treatment (TTNT), Overall Survival (OS) and safety. Patients received a median of 6 cycles and after a median follow up of 40 months, patients in the VR-CAP group demonstrated a significantly longer median PFS (25 months vs. 14 months; HR=0.63;P<0.001) with a 37% relative improvement in the PFS compared to those who were treated with standard R-CHOP. Patients in the VR-CAP group also had a higher overall response rate (88 vs 85%) and a higher rate of complete response (44% vs. 34%). The most common adverse reactions occurring in 20% or more of patients receiving the VR-CAP regimen were neutropenia, leukopenia, anemia, thrombocytopenia, lymphopenia, peripheral neuropathy, pyrexia, nausea and diarrhea. Infections were reported for 31% of patients in the VR-CAP group compared to 23% of the patients in the R-CHOP group. The authors concluded that VR-CAP significantly prolonged PFS and consistently improved secondary efficacy endpoints, compared to R-CHOP, in newly diagnosed, Bone Marrow Transplant ineligible Mantle Cell Lymphoma patients with manageable toxicity. Proteosome inhibition with a VELCADE® based chemotherapy regimen has opened the doors for more effective therapies for Mantle Cell Lymphoma patients. Cavalli F, Rooney B, Pei L, et al. J Clin Oncol 32:5s, 2014 (suppl; abstr 8500)</s
This latest approval was based on the results of an international, randomized, open-label phase III trial in which 487 patients with stage II to IV MCL, who were ineligible or not considered for Bone Marrow Transplantation, received VR-CAP (N = 243) or R-CHOP (N = 244). VR- CAP is essentially R-CHOP with the Vincristine replaced by VELCADE®. So, VR-CAP regimen consisted of VELCADE® administered IV at 1.3 mg/m2 on days 1, 4, 8, and 11, RITUXAN® (Rituximab) 375 mg/m2 IV given on day 1, Cyclophosphamide 750 mg/m2 IV on day 1, Doxorubicin 50 mg/m2 IV on day 1 and Prednisone at 100 mg/m2 PO on days 1 to 5 of a 21 day cycle for 6-8 cycles. R-CHOP regimen was exactly similar except that Vincristine 1.4 mg/m2 (max 2 mg) IV was administered on day 1 of each cycle instead of VELCADE®. The primary endpoint was Progression Free Survival (PFS) and secondary endpoints included Time To Progression (TTP), Time To Next Treatment (TTNT), Overall Survival (OS) and safety. Patients received a median of 6 cycles and after a median follow up of 40 months, patients in the VR-CAP group demonstrated a significantly longer median PFS (25 months vs. 14 months; HR=0.63;P<0.001) with a 37% relative improvement in the PFS compared to those who were treated with standard R-CHOP. Patients in the VR-CAP group also had a higher overall response rate (88 vs 85%) and a higher rate of complete response (44% vs. 34%). The most common adverse reactions occurring in 20% or more of patients receiving the VR-CAP regimen were neutropenia, leukopenia, anemia, thrombocytopenia, lymphopenia, peripheral neuropathy, pyrexia, nausea and diarrhea. Infections were reported for 31% of patients in the VR-CAP group compared to 23% of the patients in the R-CHOP group. The authors concluded that VR-CAP significantly prolonged PFS and consistently improved secondary efficacy endpoints, compared to R-CHOP, in newly diagnosed, Bone Marrow Transplant ineligible Mantle Cell Lymphoma patients with manageable toxicity. Proteosome inhibition with a VELCADE® based chemotherapy regimen has opened the doors for more effective therapies for Mantle Cell Lymphoma patients. Cavalli F, Rooney B, Pei L, et al. J Clin Oncol 32:5s, 2014 (suppl; abstr 8500)</s
Screening for Lung Cancer US Preventive Services Task Force Recommendation Statement
SUMMARY: The Centers for Medicare & Medicaid Services (CMS) on November 14, 2014, proposed that the evidence is sufficient, to add a Lung cancer screening counseling and shared decision making visit for appropriate beneficiaries. Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. It is the leading cause of cancer death among both men and women. The American Cancer Society estimates that over 224,000 new cases of lung cancer will be diagnosed in the United States in 2014 and over 159,000 will die of the disease. Given the incidence and mortality related to Lung cancer, several studies were conducted dating back to the 1960’s and 1970’s in an attempt to find an appropriate screening test for Lung cancer. They included testing sputum cytology and chest radiography or a combination of both. However, these screening methodologies did not conclusively demonstrate improvements in health outcomes. The results of a NCI-sponsored National Lung Screening Trial (NLST) published in 2011, was more optimistic. In this federally funded U.S. study, 53,439 asymptomatic participants, 55 to 74 years of age, with at least 30 pack-year smoking history were enrolled and randomized to undergo annual screening with either Low dose CT scan (n=26,715) or a chest X-Ray (n=26,724), for three years. The use of Low Dose CT (LDCT) scans for 3 years in this high risk, healthy patients, resulted in a 20% reduction in Lung cancer mortality, compared to screening with a chest X-Ray. Based on these findings, Lung cancer screening was felt appropriate for the following groups of patients:
1) People 55-74 years of age with no signs or symptoms of Lung disease or lung Cancer
2) Current or former smoker with a 30 pack year smoking history (Number of years smoked multiplied by the number of packs of cigarettes per day with each pack containing 20 cigarettes)
3) Former smokers who has quit smoking within the past 15 years
The United States Preventive Services Task Force (USPSTF) recommended annual screening for lung cancer with Low Dose Computed Tomography in adult individuals, between ages 55 to 80 years who have a 30 pack-year smoking history and currently smoke or have quit within the past 15 years. 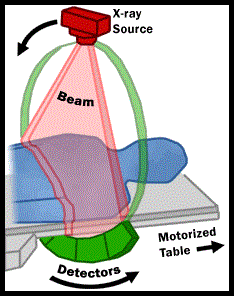 Screening should be discontinued once a person has not smoked for 15 years or develops a health problem that substantially limits life expectancy or the ability or willingness to have curative lung surgery. This was a Grade: B recommendation which meant that the USPSTF recommends the service and there is high certainty that the net benefit is moderate or there is moderate certainty that the net benefit is moderate to substantial. This therefore meant that clinicians offer or provide this service to these high risk individuals.
Screening should be discontinued once a person has not smoked for 15 years or develops a health problem that substantially limits life expectancy or the ability or willingness to have curative lung surgery. This was a Grade: B recommendation which meant that the USPSTF recommends the service and there is high certainty that the net benefit is moderate or there is moderate certainty that the net benefit is moderate to substantial. This therefore meant that clinicians offer or provide this service to these high risk individuals.
Based on this information the Centers for Medicare & Medicaid Services (CMS) on November 14, 2014, proposed that the evidence is sufficient, to add a Lung cancer screening counseling and shared decision making visit. CMS proposed, screening for Lung cancer with Low Dose Computed Tomography (LDCT), for appropriate beneficiaries, once per year, as an additional preventive service benefit under the Medicare program, only if all of the following criteria are met:
1. Age 55-74 years
2. Asymptomatic (no signs or symptoms of lung disease)
3. Tobacco smoking history of at least 30 pack-years (one pack-year = smoking one pack per day for one year; 1 pack = 20 cigarettes)
4. Current smoker or one who has quit smoking within the last 15 years
5. A lung cancer screening counseling and shared decision making visit which includes the use of one or more decision aids discussing the benefits, harms, follow-up diagnostic testing, over-diagnosis, false positive rate, and total radiation exposure
6. Counseling on the importance of adherence to annual LDCT lung cancer screening, impact of comorbidities and ability or willingness to undergo diagnosis and treatment
7. Counseling on the importance of maintaining cigarette smoking abstinence if former smoker, or smoking cessation if current smoker and, if appropriate, offering additional Medicare-covered tobacco cessation counseling services
Lung Cancer screening is performed using a non-contrast, Low Dose CT scan (LDCT) at an accredited advanced diagnostic imaging center with an effective radiation dose less than 1.5 mSv (the equivalent of 15 chest x-rays), compared to a standard chest CT with a median radiation dose of 8 mSv. The imaging center must collect and submit required data to a CMS-approved national registry for each LDCT lung cancer screening performed. Moyer VA, et al. on behalf of the U.S. Preventive Services Task Force. Ann Intern Med. 2014;160:330-338.
Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia
SUMMARY: The FDA has granted Breakthrough Therapy Designation to immunotherapy with CTL019, which are genetically engineered T-cells. Chimeric Antigen Receptor (CAR) T-cell therapy is a type of immunotherapy in which T cells collected from the patient’s own blood and are genetically engineered to produce special receptors on their surface called chimeric antigen receptors (CAR’s). The cytotoxic T cells with these chimeric antigen receptors on their surface are now able to recognize a specific antigen on tumor cells. These engineered CAR T-cells which are grown in the lab are then infused into the patient and they in turn proliferate in the patient’s body and the engineered receptor on their surface help recognize and kill cancer cells that expresses that specific antigen. CTL019 are genetically engineered T-cells using CAR technology that seeks out cancer cells expressing the antigen CD19, which is found uniquely on B cells and destroy them.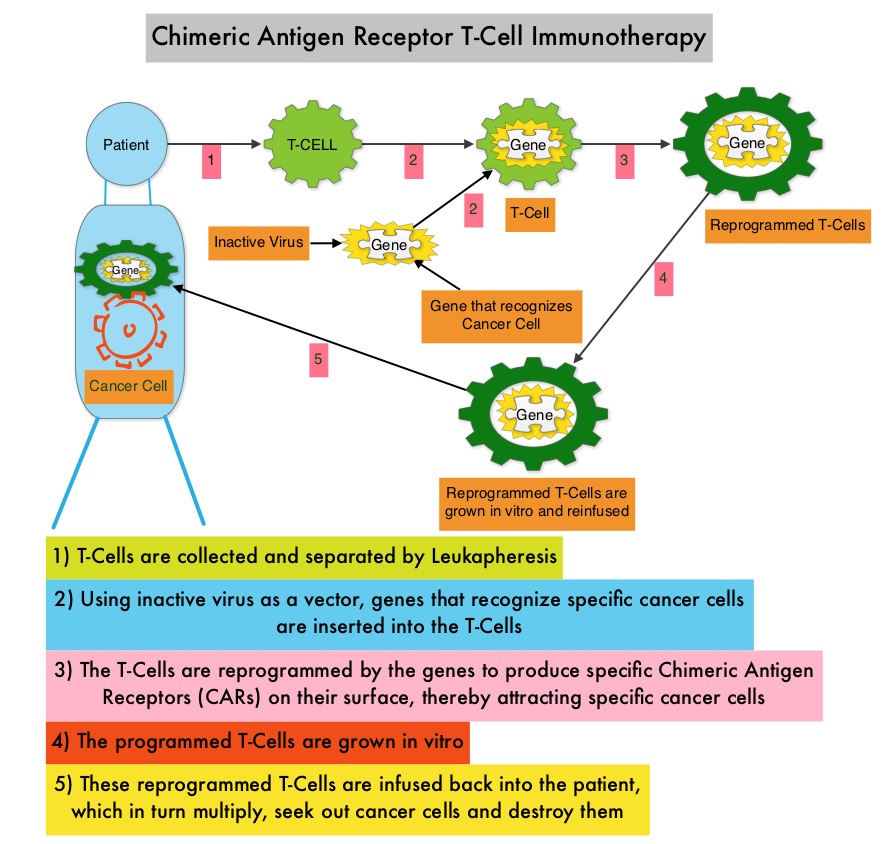 Patients, following treatment with CAR T-cells, develop B-cell aplasia (absence of CD19 positive cells) due to B-cell destruction and may need immunoglobin replacement. Hence, B-cell aplasia can be a useful therapeutic marker, as continued B-cell aplasia has been seen in all patients who had sustained remission, following CAR T-cell therapy. Cytokine Release Syndrome, an inflammatory process is the most common and serious side effect of CAR T-cell therapy and is associated with marked elevation of Interleukin-6. Cytokine release is important for T-cell activation and can result in high fevers and myalgias. This is usually self limiting although if severe can be associated with hypotension and respiratory insufficiency. Tocilizumab, an Interleukin-6 receptor blocking antibody produces a rapid improvement in symptoms. This is however not recommended unless the symptoms are severe and life threatening, as blunting the cytokine response can in turn negate T-cell proliferation. Elevated serum Ferritin and C-reactive protein levels are surrogate markers for severe Cytokine Release Syndrome. The CAR T-cells have been shown to also access sanctuary sites such as the central nervous system and eradicate cancer cells. CD19 antigen is expressed by majority of the B cell malignancies and therefore most studies using CAR T-cell therapy have focused on the treatment of advanced B-cell malignancies such as Chronic Lymphocytic Leukemia (CLL), Acute Lymphoblastic Leukemia (ALL) and Non Hodgkin lymphoma (NHL), such as Diffuse Large B-Cell Lymphoma (DLBCL). Previously published studies have shown significant responses with CAR T-cell therapy in patients with relapsed and refractory B-cell ALL. But the durability of remission has remained unclear.
Patients, following treatment with CAR T-cells, develop B-cell aplasia (absence of CD19 positive cells) due to B-cell destruction and may need immunoglobin replacement. Hence, B-cell aplasia can be a useful therapeutic marker, as continued B-cell aplasia has been seen in all patients who had sustained remission, following CAR T-cell therapy. Cytokine Release Syndrome, an inflammatory process is the most common and serious side effect of CAR T-cell therapy and is associated with marked elevation of Interleukin-6. Cytokine release is important for T-cell activation and can result in high fevers and myalgias. This is usually self limiting although if severe can be associated with hypotension and respiratory insufficiency. Tocilizumab, an Interleukin-6 receptor blocking antibody produces a rapid improvement in symptoms. This is however not recommended unless the symptoms are severe and life threatening, as blunting the cytokine response can in turn negate T-cell proliferation. Elevated serum Ferritin and C-reactive protein levels are surrogate markers for severe Cytokine Release Syndrome. The CAR T-cells have been shown to also access sanctuary sites such as the central nervous system and eradicate cancer cells. CD19 antigen is expressed by majority of the B cell malignancies and therefore most studies using CAR T-cell therapy have focused on the treatment of advanced B-cell malignancies such as Chronic Lymphocytic Leukemia (CLL), Acute Lymphoblastic Leukemia (ALL) and Non Hodgkin lymphoma (NHL), such as Diffuse Large B-Cell Lymphoma (DLBCL). Previously published studies have shown significant responses with CAR T-cell therapy in patients with relapsed and refractory B-cell ALL. But the durability of remission has remained unclear.
The authors in this study, treated a total of 30 patients with relapsed or refractory ALL ( included those who had relapsed after allogeneic stem cell transplantation and those refractory to CD19 directed bispecific antibody Blinatumomab), with autologous Chimeric Antigen Receptor (CAR) T-cells (CTL019 T-cells) and monitored response rates, toxicities as well as proliferation and persistence of circulating CTL019 T-cells in the patient’s body. The first assessment was performed 1 month after infusion of CTL019 and 90% of the patients were in complete remission and sustained remissions were noted for up to 2 years. At a median follow up of 6 months, the event free survival was 67% and overall survival was 78%. The authors compared this efficacy data with the FDA approved agents for relapsed ALL such as Clofarabine, Nelarabine and Liposomal encapsulated Vincristine, which have a complete remission of less than 25% with a median duration of response of 4-9 weeks. Persisting CTL019 T-cells in the body is a marker of therapeutic efficacy. CTL019 T-cells proliferated in the patient’s body and was detectable in the blood bone marrow, and cerebrospinal fluid of patients who had a response. At 6 months, the probability that a patient would have persistence of CTL019 T-cells was 68% and the probability that a patient would have relapse free B-cell aplasia was 73%. Severe Cytokine Release Syndrome was noted in 27% of the patients and these patients had a higher disease burden before CTL019 infusion. All of these patients were effectively treated with the Interleukin-6 receptor blocking antibody Tocilizumab. The authors concluded that Chimeric Antigen Receptor modified T-cell therapy against CD19 positive cells (CTL019) was highly efficacious, in patients with relapsed and refractory ALL and was associated with a high and durable remission rate. This technology may be applied to other malignancies, as new antigen targets are identified. Maude SL, Frey N, Shaw PA, et al. N Engl J Med 2014; 371:1507-1517
Bevacizumab Combined With Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer The AURELIA Open-Label Randomized Phase III Trial
SUMMARY:The FDA recently approved AVASTIN® (Bevacizumab) in combination with chemotherapy for the treatment of patients with platinum-resistant, recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer. It is estimated that in the United States, approximately 22,000 women will be diagnosed with ovarian cancer in 2014 and a little over 14,000 women will die of the disease. In spite of significantly improved median survival following aggressive surgical debulking and platinum plus taxane based therapy, long term cure rate is approximately 20-30%. Majority of the patients relapse in 18-24 months and 25% of these patients are Platinum Resistant. These platinum resistant patients are usually treated with single agent chemotherapy drugs such as DOXIL® (Pegylated Liposomal Doxorubicin-PLD), TAXOL® (Paclitaxel) and HYCAMTIN® (Topotecan), with an expected response rate of 10-15%, median response duration of about 3-4 months and median Overall Survival of approximately 12 months. AURELIA (Avastin Use in Platinum-Resistant Epithelial Ovarian Cancer) is a multicenter, randomized, open-label, Phase III study in which 361 women with platinum resistant recurrent epithelial ovarian, primary peritoneal or fallopian tube cancer were enrolled. These patients had disease progression within six months of their platinum based chemotherapy (Platinum Resistant) and were randomly assigned to receive AVASTIN® (Bevacizumab) 10 mg/kg every 2 weeks or 15 mg/kg every 3 weeks in combination with investigators choice of single agent chemotherapy agent (N=179) or single agent chemotherapy alone (N=182). Chemotherapy included one of the following agents – TAXOL® 80 mg/m2 on days 1, 8, 15 and 22 every 4 weeks, DOXIL® 40 mg/m2 on day 1 every 4 weeks or HYCAMTIN® either 4 mg/m2 on days 1, 8 and 15 every 4 weeks or 1.25 mg/m2 on days 1-5 every 3 weeks. Patients with refractory disease, history of bowel obstruction, or those who had received two or more prior anticancer regimens were excluded. Treatment was given until disease progression. Patients who had progressed on single agent chemotherapy were allowed to cross over to AVASTIN® group. 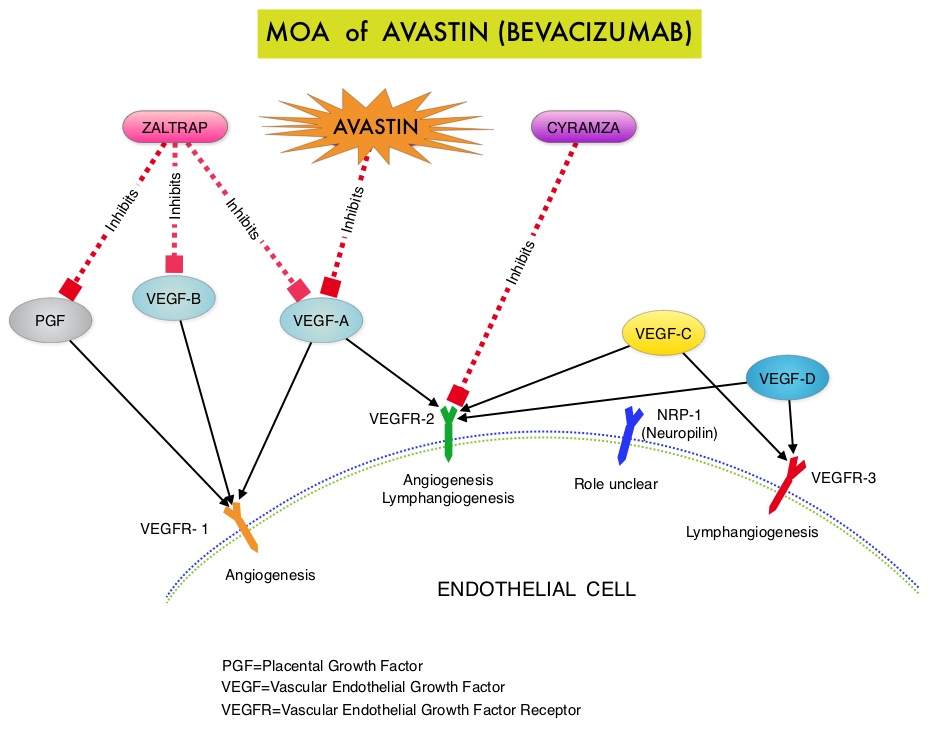 The primary end point was Progression Free Survival (PFS) and secondary end points included Objective Response Rate (ORR), Overall Survival (OS), safety, and patient reported outcomes. The combination of AVASTIN® plus chemotherapy resulted in a 62% reduction in the risk of progression compared to those who received chemotherapy alone, with a median PFS of 6.8 months for the AVASTIN® plus chemotherapy group versus 3.4 months for the single agent chemotherapy group (HR=0.38, P<0.0001) and thus met the primary endpoint of this clinical trial. This PFS benefit was seen consistently across all subgroups including the subgroup of patients with ascites. The ORR was 27.3% with the AVASTIN® combination versus 11.8% with single agent chemotherapy (P =0.001). The median OS was 16.6 months for the AVASTIN® combination versus 13.3 months for the single agent chemotherapy group (HR=0.85; P < .17). The lack of statistical significance in the OS has been attributed to cross over of 40% of patients, initially randomized to the chemotherapy alone group, who upon progression received AVASTIN®. There was a 15% improvement in abdominal and GI symptoms as reported by patients, with the AVASTIN® combination, compared to chemotherapy alone. On exploratory analyses it was noted that the addition of AVASTIN® to TAXOL® resulted in the most benefit, with a 5.7 month improvement in median PFS (9.6 versus 3.9 months), a 23% improvement in the overall response rate (53% versus 30%) and a 9.2 month improvement in median OS (22.4 versus 13.2 months) compared to single agent TAXOL®. This benefit was seen in spite of the fact that 97% of the patients in the TAXOL® group had received this agent with previous chemotherapy regimens. These findings suggest that patients who have received prior treatment with TAXOL® may benefit from AVASTIN® plus weekly TAXOL®. The most common adverse reactions (greater than or equal to 15%) in patients treated with AVASTIN® plus chemotherapy were neutropenia, peripheral neuropathy, hypertension and GI perforation occurred in 1.7% of these patients. This low perforation rate has been attributed to the exclusion of patients with rectosigmoid involvement by pelvic examination or bowel involvement on CT scan as well as those with clinical symptoms of bowel obstruction. The authors concluded that AVASTIN® in combination with chemotherapy significantly improved Progression Free Survival and Objective Response Rates in patients with Platinum Resistant Recurrent Ovarian Cancer. Pujade-Lauraine E, Hilpert F, Weber B, et al. J Clin Oncol 2014;32:1302-1308
The primary end point was Progression Free Survival (PFS) and secondary end points included Objective Response Rate (ORR), Overall Survival (OS), safety, and patient reported outcomes. The combination of AVASTIN® plus chemotherapy resulted in a 62% reduction in the risk of progression compared to those who received chemotherapy alone, with a median PFS of 6.8 months for the AVASTIN® plus chemotherapy group versus 3.4 months for the single agent chemotherapy group (HR=0.38, P<0.0001) and thus met the primary endpoint of this clinical trial. This PFS benefit was seen consistently across all subgroups including the subgroup of patients with ascites. The ORR was 27.3% with the AVASTIN® combination versus 11.8% with single agent chemotherapy (P =0.001). The median OS was 16.6 months for the AVASTIN® combination versus 13.3 months for the single agent chemotherapy group (HR=0.85; P < .17). The lack of statistical significance in the OS has been attributed to cross over of 40% of patients, initially randomized to the chemotherapy alone group, who upon progression received AVASTIN®. There was a 15% improvement in abdominal and GI symptoms as reported by patients, with the AVASTIN® combination, compared to chemotherapy alone. On exploratory analyses it was noted that the addition of AVASTIN® to TAXOL® resulted in the most benefit, with a 5.7 month improvement in median PFS (9.6 versus 3.9 months), a 23% improvement in the overall response rate (53% versus 30%) and a 9.2 month improvement in median OS (22.4 versus 13.2 months) compared to single agent TAXOL®. This benefit was seen in spite of the fact that 97% of the patients in the TAXOL® group had received this agent with previous chemotherapy regimens. These findings suggest that patients who have received prior treatment with TAXOL® may benefit from AVASTIN® plus weekly TAXOL®. The most common adverse reactions (greater than or equal to 15%) in patients treated with AVASTIN® plus chemotherapy were neutropenia, peripheral neuropathy, hypertension and GI perforation occurred in 1.7% of these patients. This low perforation rate has been attributed to the exclusion of patients with rectosigmoid involvement by pelvic examination or bowel involvement on CT scan as well as those with clinical symptoms of bowel obstruction. The authors concluded that AVASTIN® in combination with chemotherapy significantly improved Progression Free Survival and Objective Response Rates in patients with Platinum Resistant Recurrent Ovarian Cancer. Pujade-Lauraine E, Hilpert F, Weber B, et al. J Clin Oncol 2014;32:1302-1308
CYRAMZA® (Ramucirumab)
The FDA on November 5, 2014 approved CYRAMZA® for use in combination with TAXOL® (Paclitaxel), for the treatment of patients with advanced Gastric or GastroEsophageal Junction (GEJ) adenocarcinoma. CYRAMZA® was approved in April, 2014 as a single agent for the treatment of patients with advanced Gastric or GEJ adenocarcinoma refractory to or progressive following first-line therapy with platinum or fluoropyrimidine chemotherapy. CYRAMZA® injection for intravenous infusion is a product of Eli Lilly and Company.
Ipilimumab Plus Sargramostim vs Ipilimumab Alone for Treatment of Metastatic Melanoma – A Randomized Clinical Trial
SUMMARY: It is estimated that in the US, approximately 76,000 new cases of melanoma will be diagnosed and close to 8000 individuals will die of the disease in 2014. The incidence of melanoma has been on the rise for the past three decades. Unlike other malignancies, the role of chemotherapy for the treatment of melanoma has been limited. Treatment of advanced melanoma with immunotherapy using a cytokine, Interleukin-2 (IL-2) produced by T cells during an immune response, was first explored in the mid 1970’s. Durable responses were noted in a very small percentage of patients but this was associated with significant toxicities. This however opened the doors for the development a novel immunotherapeutic approaches, with a better understanding of the Immune checkpoints.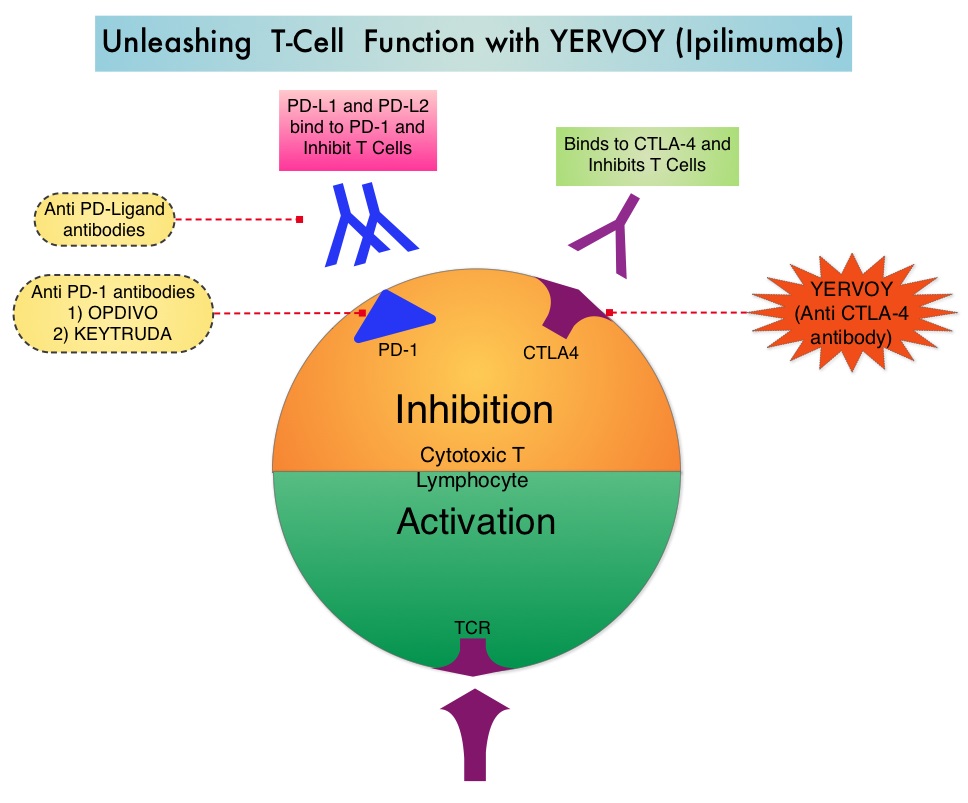 Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. The authors in this randomized study, compared the efficacy of YERVOY® (Ipilimumab) plus Sargramostim with YERVOY® alone, for treatment of metastatic melanoma. The rationale for this study was based on the synergy that was noted between YERVOY® and GM-CSF in preclinical models. The first immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® is a fully human IgG1monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA- 4 and counteracts immune regulatory cells. YERVOY® has been shown to prolong overall survival in patients with previously treated, unresectable or metastatic melanoma. GM-CSF is a cytokine that enhances the antitumor activity of T and B lymphocytes by activating the antigen presenting dendritic cells and recruiting macrophages. It however can induce negative regulatory immune responses.
Immune checkpoints are cell surface inhibitory proteins/receptors that are expressed on activated T cells. They harness the immune system and prevent uncontrolled immune reactions. Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte activation. The T cells of the immune system therefore play a very important role in modulating the immune system. Under normal circumstances, inhibition of an intense immune response and switching off the T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. With the recognition of Immune checkpoint proteins and their role in suppressing antitumor immunity, antibodies are being developed that target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152), PD-1(Programmed cell Death 1), etc. By doing so, one would expect to unleash the T cells, resulting in T cell proliferation, activation and a therapeutic response. The authors in this randomized study, compared the efficacy of YERVOY® (Ipilimumab) plus Sargramostim with YERVOY® alone, for treatment of metastatic melanoma. The rationale for this study was based on the synergy that was noted between YERVOY® and GM-CSF in preclinical models. The first immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® is a fully human IgG1monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA- 4 and counteracts immune regulatory cells. YERVOY® has been shown to prolong overall survival in patients with previously treated, unresectable or metastatic melanoma. GM-CSF is a cytokine that enhances the antitumor activity of T and B lymphocytes by activating the antigen presenting dendritic cells and recruiting macrophages. It however can induce negative regulatory immune responses.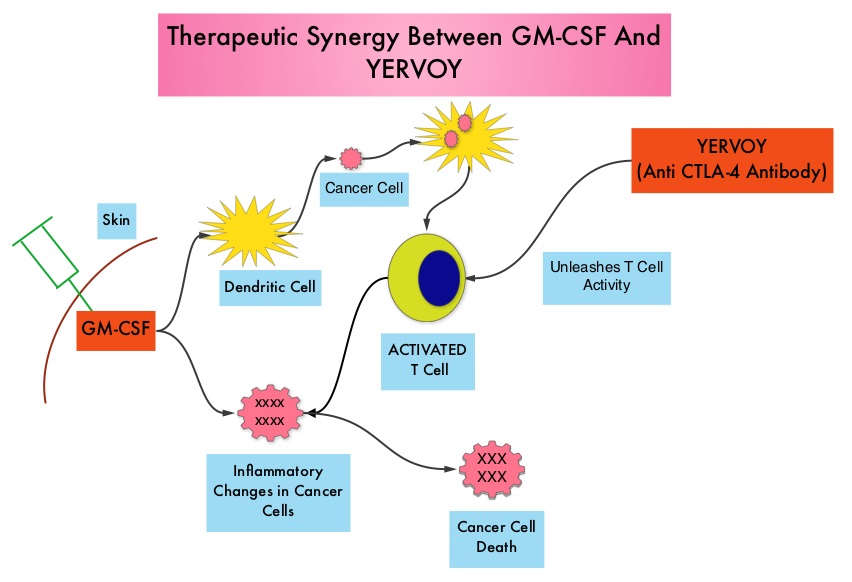 In this phase II randomized clinical trial conducted by the Eastern Cooperative Oncology Group (ECOG), patients with unresectable stage III or IV melanoma (N = 245), who had received at least 1 prior therapy and with no central nervous system metastases were randomized to receive either YERVOY® along with Sargramostim (N=123) or YERVOY® alone (N=122). Patients in the combination group (Group A) received YERVOY®10 mg/kg, IV on day 1 along with Sargramostim 250 μg given subcutaneously, on days 1 thru 14 of a 21day cycle, every 3 weeks for four cycles followed by YERVOY® maintenance every 12 weeks. Patients in Group B received YERVOY® alone. Treatment was continued until disease progression or uncontrolled toxicities. The primary endpoint was comparison of length of Overall Survival (OS). Secondary end points included Progression Free Survival (PFS), response rate, safety, and tolerability. With a median follow up of 13.3 months, the median OS for the combination of YERVOY® plus Sargramostim was 17.5 months vs 12.7 months for YERVOY® alone. The one year survival rate for YERVOY® plus Sargramostim was 68.9% compared to 52.9% for YERVOY® alone (HR=0.64; P=0.01). The median PFS was similar and was 3.1 months in both study groups. The explanation for similar PFS in both treatment groups may be due to both YERVOY® and Sargramostim bringing about inflammatory changes at the tumor sites, which in turn could be misinterpreted as disease progression, on radiological studies. The authors commented that PFS may not be an appropriate endpoint in immunotherapy trials. Grade 3 to 5 adverse events were less in the combination group (44.9%) compared to 58% for single agent YERVOY® (P=0.04). The authors concluded that treatment of unresectable stage III or IV melanoma patients with YERVOY® plus Sargramostim resulted in significantly longer overall survival with lower toxicities, compared to YERVOY® alone. Hodi SF, Lee S, McDermott DF, et al. JAMA 2014;312:1744-1753
In this phase II randomized clinical trial conducted by the Eastern Cooperative Oncology Group (ECOG), patients with unresectable stage III or IV melanoma (N = 245), who had received at least 1 prior therapy and with no central nervous system metastases were randomized to receive either YERVOY® along with Sargramostim (N=123) or YERVOY® alone (N=122). Patients in the combination group (Group A) received YERVOY®10 mg/kg, IV on day 1 along with Sargramostim 250 μg given subcutaneously, on days 1 thru 14 of a 21day cycle, every 3 weeks for four cycles followed by YERVOY® maintenance every 12 weeks. Patients in Group B received YERVOY® alone. Treatment was continued until disease progression or uncontrolled toxicities. The primary endpoint was comparison of length of Overall Survival (OS). Secondary end points included Progression Free Survival (PFS), response rate, safety, and tolerability. With a median follow up of 13.3 months, the median OS for the combination of YERVOY® plus Sargramostim was 17.5 months vs 12.7 months for YERVOY® alone. The one year survival rate for YERVOY® plus Sargramostim was 68.9% compared to 52.9% for YERVOY® alone (HR=0.64; P=0.01). The median PFS was similar and was 3.1 months in both study groups. The explanation for similar PFS in both treatment groups may be due to both YERVOY® and Sargramostim bringing about inflammatory changes at the tumor sites, which in turn could be misinterpreted as disease progression, on radiological studies. The authors commented that PFS may not be an appropriate endpoint in immunotherapy trials. Grade 3 to 5 adverse events were less in the combination group (44.9%) compared to 58% for single agent YERVOY® (P=0.04). The authors concluded that treatment of unresectable stage III or IV melanoma patients with YERVOY® plus Sargramostim resulted in significantly longer overall survival with lower toxicities, compared to YERVOY® alone. Hodi SF, Lee S, McDermott DF, et al. JAMA 2014;312:1744-1753