
SUMMARY: The role of Somatostatin analogs such as SANDOSTATIN® (Octreotide) for symptom control in patients with gastrointestinal and pancreatic NeuroEndocrine Tumors (NETs) is well established. SANDOSTATIN® also demonstrated antiproliferative activity in controlling tumor growth of well-differentiated metastatic midgut NETs (Carcinoid), by lengthening the Time to Tumor Progression (TTP), when compared with placebo (PROMID Study). Whether SANDOSTATIN® prolongs Overall Survival (OS) remained unclear. The study investigators now reported the long term follow up data from the same PROMID trial. Between 2001 and 2008, 85 patients were randomly assigned to receive either SANDOSTATIN® LAR (N=42) or Placebo (N=43). On disease progression, patients in the placebo group were allowed to crossover and receive SANDOSTATIN® LAR. Outcomes in patients with Hepatic tumor Load (HL – percentage of liver replaced by malignancy) at study entry of 10% or less, was compared to those whose HL was more than 10%. The median OS by January 2013 in the Placebo arm was 84 months whereas the median OS in the SANDOSTATIN® LAR group was not reached, suggesting that the OS in this group will exceed 84 months and therefore a longer follow up would be needed. Patients with HL 10% or less benefited the most whereas those with high HL did not have OS benefit with SANDOSTATIN® LAR. The authors concluded that SANDOSTATIN® LAR prolongs TTP as well as OS in patients with metastatic midgut NETs, carrying a Hepatic Load of 10% or less. Arnold R, Wittenberg M, Rinke A, et al. J Clin Oncol 31, 2013 (suppl; abstr 4030)
Author: RR
Baseline Selenium Status and Effects of Selenium and Vitamin E Supplementation on Prostate Cancer Risk
SUMMARY: Selenium and Vitamin E Cancer Prevention Trial (SELECT), is a multicenter, randomized, placebo-controlled trial, conducted by the SWOG cooperative group, that involved more than 35,000 men. Participants were randomized to receive either, a) Selenium and Vitamin E, b) Selenium and a placebo, c) Vitamin E and a placebo or d) Two placebos. The purpose of this trial was to determine if high dose vitamin E (400 IU/day) and/or Selenium (200 mcg/day) supplements could decrease the incidence of prostate cancer. The level/concentration of Selenium in participants toenail clippings was measured at the time of study participation and the goal was to also determine whether Selenium supplements would benefit the subset of participants with low Selenium levels at baseline. Both Vitamin E and Selenium are antioxidants and Vitamin E rich foods include vegetables, vegetable oils, nuts, and egg yolks whereas Selenium a nonmetallic trace element is found in rice, wheat, seafood, meat, and Brazil nuts. 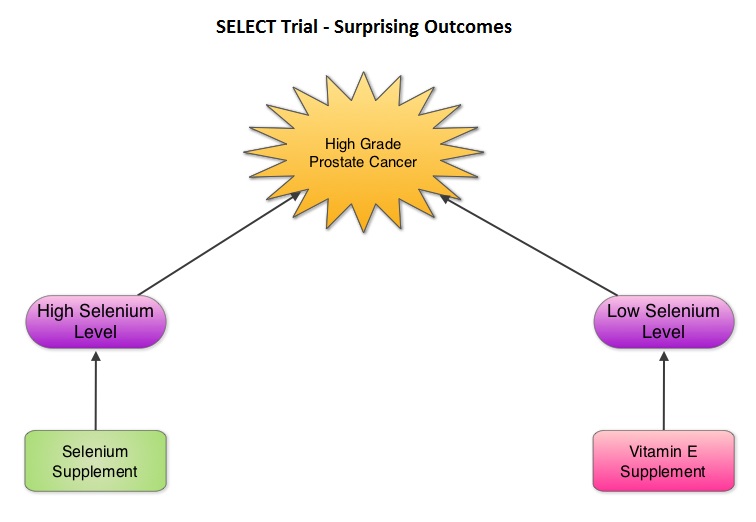 The SELECT trial, which began in 2001, was stopped early in 2008, as Selenium and Vitamin E, taken alone or together for an average of five and a half years did not decrease the incidence of prostate cancer. In 2011, an update on the SELECT trial data suggested that men who were randomized to the vitamin E alone had a 17 percent increased risk of prostate cancer compared to those men taking placebo. The authors in this case–cohort study continued follow up of the SELECT trial participants and with the Selenium levels data from toenail clippings, compared the effect of Selenium and Vitamin E, taken either alone or together, on the risk of prostate cancer, among 1739 men who were diagnosed with prostate cancer, of whom 489 participants developed high-grade prostate cancer. The control group for comparison was a random sample of 3117 men without prostate cancer and they were matched to the cases by race and age. It was noted that an individual’s baseline Selenium level, in the absence of supplementation, was not associated with prostate cancer risk. However, in men who had high baseline Selenium levels, Selenium supplements almost doubled (91%) the risk of high grade prostate cancer (P=0.007). Conversely, Vitamin E supplements had no effect among men with high baseline Selenium levels but doubled the risk of high grade prostate cancer among men with low baseline Selenium levels. Frankel et al. in an accompanying editorial point out that the dose of Vitamin E in the SELECT trial was significantly higher (400 IU/day) than the dose that was selected in the Alpha-Tocopherol Beta Carotene (ATBC) Cancer Prevention trial (50 IU/day), a study that was designed to test Vitamin E and beta carotene for lung cancer prevention in smokers. In the ATBC trial, a decrease in the incidence of prostate cancer incidence was observed, although this was a secondary finding and this study was not designed to determine prostate cancer risk. They comment that high doses of Vitamin E (Alpha-Tocopherol), suppresses the more potentially beneficial serum Gamma-Tocopherol which is the prevalent dietary form of Vitamin E in the United States. Selenium deficiency in the U.S. is not common and any benefit with Selenium supplements can only be seen in those who are Selenium deficient and high doses may be detrimental. The authors concluded that in the SELECT trial, the combination of both Vitamin E and Selenium did not reduce the risk of prostate cancer or any other cancer or heart disease and was in fact harmful for a significant number of individuals. Therefore, men 55 years of age or more should avoid Vitamin E or Selenium supplements at doses that exceed the recommended dietary intake. Kristal AR, Darke AK, Morris JS, et al. J Natl Cancer Inst; First published online 22 February 2014, doi: 10.1093/jnci/djt456
The SELECT trial, which began in 2001, was stopped early in 2008, as Selenium and Vitamin E, taken alone or together for an average of five and a half years did not decrease the incidence of prostate cancer. In 2011, an update on the SELECT trial data suggested that men who were randomized to the vitamin E alone had a 17 percent increased risk of prostate cancer compared to those men taking placebo. The authors in this case–cohort study continued follow up of the SELECT trial participants and with the Selenium levels data from toenail clippings, compared the effect of Selenium and Vitamin E, taken either alone or together, on the risk of prostate cancer, among 1739 men who were diagnosed with prostate cancer, of whom 489 participants developed high-grade prostate cancer. The control group for comparison was a random sample of 3117 men without prostate cancer and they were matched to the cases by race and age. It was noted that an individual’s baseline Selenium level, in the absence of supplementation, was not associated with prostate cancer risk. However, in men who had high baseline Selenium levels, Selenium supplements almost doubled (91%) the risk of high grade prostate cancer (P=0.007). Conversely, Vitamin E supplements had no effect among men with high baseline Selenium levels but doubled the risk of high grade prostate cancer among men with low baseline Selenium levels. Frankel et al. in an accompanying editorial point out that the dose of Vitamin E in the SELECT trial was significantly higher (400 IU/day) than the dose that was selected in the Alpha-Tocopherol Beta Carotene (ATBC) Cancer Prevention trial (50 IU/day), a study that was designed to test Vitamin E and beta carotene for lung cancer prevention in smokers. In the ATBC trial, a decrease in the incidence of prostate cancer incidence was observed, although this was a secondary finding and this study was not designed to determine prostate cancer risk. They comment that high doses of Vitamin E (Alpha-Tocopherol), suppresses the more potentially beneficial serum Gamma-Tocopherol which is the prevalent dietary form of Vitamin E in the United States. Selenium deficiency in the U.S. is not common and any benefit with Selenium supplements can only be seen in those who are Selenium deficient and high doses may be detrimental. The authors concluded that in the SELECT trial, the combination of both Vitamin E and Selenium did not reduce the risk of prostate cancer or any other cancer or heart disease and was in fact harmful for a significant number of individuals. Therefore, men 55 years of age or more should avoid Vitamin E or Selenium supplements at doses that exceed the recommended dietary intake. Kristal AR, Darke AK, Morris JS, et al. J Natl Cancer Inst; First published online 22 February 2014, doi: 10.1093/jnci/djt456
Oral rivaroxaban versus standard therapy for the treatment of symptomatic venous thromboembolism a pooled analysis of the EINSTEIN-DVT and PE randomized studies
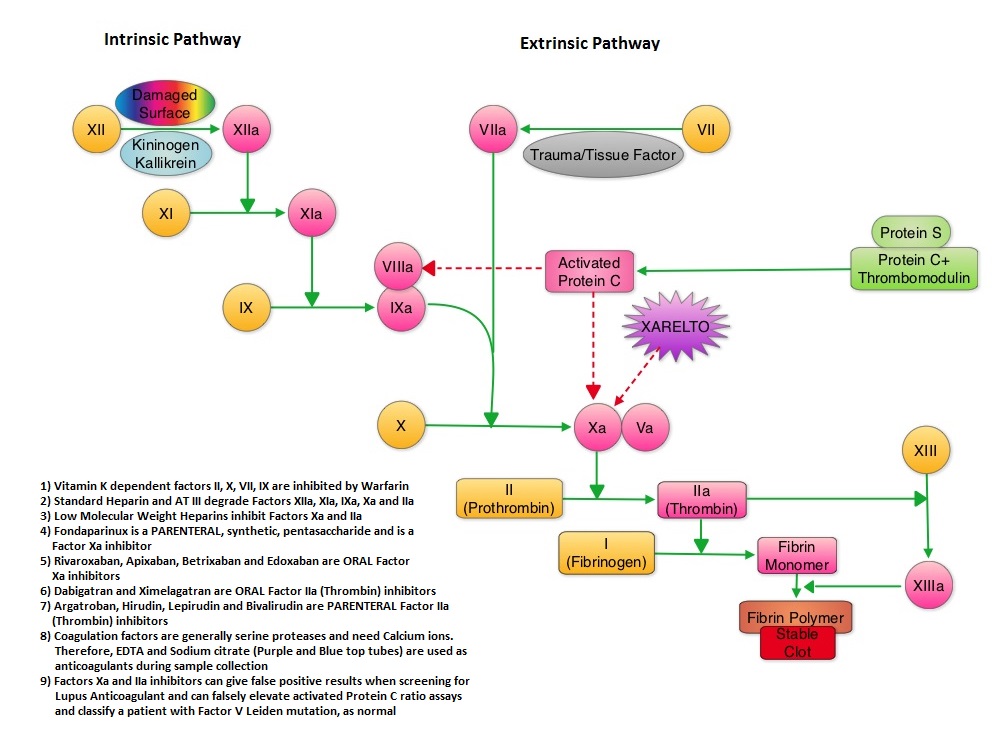
SUMMARY: XARELTO® (Rivaroxaban) is an oral, direct inhibitor of Factor Xa. Two previously published randomized studies concluded that XARELTO® was noninferior to the standard anticoagulation therapy (administration of heparin overlapped and followed by Vitamin K Antagonist), for most patients with Venous ThromboEmbolism. Further, XARELTO® in these studies also had a better safety profile compared to standard anticoagulant therapy. The authors in this pooled analysis combined the data from 2 identically designed studies, EINSTEIN-DVT and EINSTEIN-PE in which XARELTO® was compared to standard anticoagulation therapy, in patients with DVT and PE respectively. The goal of this study was to provide a more accurate estimate of the efficacy and safety of XARELTO® in elderly patients and cancer patients in whom Vitamin K Antagonists (VKA) such as COUMADIN® (Warfarin) can be associated with a higher complication rate. In addition, the study included analysis of outcomes with XARELTO® in patients with previous VTE and in those presenting with extensive thrombosis. Of the 8282 patients with VTE randomized in the pooled analysis, 4151 patients received XARELTO® and 4131 patients received LOVENOX® (Enoxaparin) / Vitamin K Antagonists. In both pooled studies, patients in the XARELTO® group received a dose of 15 mg PO BID for 21 days, followed by 20 mg PO QD whereas patients assigned to the standard anticoagulation therapy group received LOVENOX® subcutaneous at a dose of 1.0 mg/kg BID and either oral Warfarin or Acenocoumarol started within 48 hours after randomization and patients continued treatment for 3, 6, or 12 months, as determined by the local Health Care Providers. INR was closely monitored and maintained between 2-3. On final review, the analysis suggested that XARELTO® resulted in efficacy similar to standard anticoagulation therapy, with a noninferiority P<0.001. The pre-specified principal safety outcome was clinically relevant bleeding and major bleeding was observed in 40 patients belonging to the XARELTO® group and in 72 patients belonging to the standard anticoagulation therapy group (HR=0.54; P= 0.002). Similar benefits in the efficacy and safety were seen in the key subgroups evaluated, which included elderly fragile patients, cancer patients, patients presenting with extensive thrombosis and those with a history of recurrent VTE. The authors concluded that the incidence of major bleeding with XARELTO® was significantly less, particularly in the high risk group of patients, when compared to standard anticoagulation therapy and may therefore have a safety advantage, without compromising efficacy. Prins MH, Lensing AW, Bauersachs R, et al. Thrombosis Journal 2013;11:21
Improved Survival with Bevacizumab in Advanced Cervical Cancer
SUMMARY: Cervical cancer is the fourth most common cancer affecting women, worldwide. It is also the fourth most common cause of cancer death. With the availability of widespread screening techniques and HPV vaccination in the U.S., the incidence of cervical cancer is declining. Treatment of advanced cervical cancer continues to be a challenge. To address this further, the authors in this study evaluated the benefit of adding AVASTIN® (Bevacizumab) to conventional chemotherapy regimens, for patients with advanced cervical cancer. AVASTIN®, a humanized Vascular Endothelial Growth Factor (VEGF) targeted monoclonal antibody, has demonstrated single-agent activity in heavily pretreated patients with recurrent cervical carcinoma in phase II trials. In this randomized study, 452 enrolled patients with metastatic, persistent or recurrent cervical cancer received one of the four treatment regimens using a 2×2 factorial design. The four treatment groups included a) Cisplatin 50 mg/m2 plus TAXOL® (Paclitaxel) 135 or 175 mg/m2 (N=114), b) HYCAMTIN® (Topotecan) 0.75 mg/m2 given on D1 thru D3 plus TAXOL® 175 mg/m2 given on Day 1 (N=111), c) Cisplatin 50 mg/m2 plus TAXOL® 135 or 175 mg/m2 given along with AVASTIN® 15mg/kg on Day 1 (N=115) and d) HYCAMTIN® 0.75 mg/m2 given on D1 thru D3 plus TAXOL® 175 mg/m2 given on Day 1, along with AVASTIN® 15mg/kg on day 1 (N=112). Treatment was given every 21 days until disease progression, the development of unacceptable toxicities or a complete response was noted. The primary end point was Overall Survival and secondary endpoints included Progression Free Survival (PFS) and Response Rate (RR). When outcomes were analyzed, HYCAMTIN® based chemotherapy was not superior to Cisplatin based chemotherapy, regardless of prior exposure to Cisplatin. The addition of AVASTIN® to chemotherapy resulted in a significantly longer median Overall Survival (17 vs 13.3 months; HR=0.71; P=0.004), significantly longer median PFS (8.2 vs 5.9 months; HR=0.67; P=0.002) and RR (48% vs 36%; P=0.008), compared to combination chemotherapy alone. The benefit with added AVASTIN® was noted in all subgroups regardless of age, race, performance status and prior platinum exposure. Treatment was in general well tolerated without significant reduction in quality of life. As was seen in other tumor types, AVASTIN® based chemotherapy regimen was associated with a higher incidence of hypertension and thromboembolic events. The authors concluded that the addition of AVASTIN® to combination chemotherapy significantly decreased the risk of death in patients with recurrent, persistent, or metastatic cervical cancer. Tewari KS, Sill MW, Long HJ, et al. N Engl J Med 2014; 370:734-743
Impact of More Restrictive Blood Transfusion Strategies on Clinical Outcomes A Meta-analysis and Systematic Review
SUMMARY: The traditional hemoglobin trigger to recommend blood transfusions for majority of the Health Care Providers is between 7.5 and 9 g/dl. The clinical rationale is based on the premise that increasing Hgb levels increases blood oxygen content and possible oxygen delivery to the tissues. However, there are no randomized trials validating improved oxygen delivery to tissues or better clinical outcomes in any setting at this hemoglobin transfusion trigger. The authors in this provocative study conducted a comprehensive research and performed a Primary and Secondary meta-analysis. In their primary meta-analysis, they reviewed the pooled data from 3 randomized clinical trials with 2364 patients and in these trials, a less than 7g/dl hemoglobin as a transfusion trigger (restrictive transfusion strategy) was compared with a more liberal transfusion strategy and outcomes were evaluated. These endpoints included mortality, acute coronary syndrome, pulmonary edema, infections and re-bleeding risk. The combined data from these 3 trials showed that a restrictive transfusion strategy resulted in a 26% mortality reduction in hospitalized patients, 20% reduction in total mortality, 36% reduction in the risk of re-bleeding, 56% reduction in acute coronary syndrome, 52% reduction in the incidence of pulmonary edema and 14% reduction in bacterial infections, compared with a more liberal transfusion strategy. The secondary meta-analysis evaluated patients in 16 trials (these were excluded from the primary meta-analysis) that used a less restrictive transfusion trigger (hemoglobin transfusion triggers of 7.5-10g/dl) and the authors noted that outcomes were not improved with a more liberal transfusion strategy. Further it was also noted that several observational studies have shown that Hgb levels of 5-6g/dl was well tolerated in normovolemic patients without effecting oxygen delivery. Contrary to clinical presumptions, these counter-intuitive findings can be explained based on sound physiologic principles. Normovolemic hemodilution following administration of crystalloid or colloid solutions, to replace blood loss, has been associated with a reduction in systemic vascular resistance, increase in cardiac output, coronary and cerebral blood flow and synthesis of 2,3-diphosphoglycerate in red blood cells thus maintaining movement of oxygen from red blood cells to body tissues. Liberal blood transfusions may in fact impair oxygen uptake by vital tissues by increasing the blood viscosity and the resulting loss of RBC function during preservation and storage of blood. Studies have also shown that in patients with gastrointestinal bleeding, restrictive transfusion strategy results in a lower portal blood pressure and less recurrent bleeding, as higher blood pressures might disrupt a thrombus plug. The authors following this clinically relevant meta-analysis concluded that restrictive transfusion strategy resulted in better outcomes and transfusion triggers should be evidence based. Salpeter SR, Buckley JS and Chatterjee S. The American Journal of Medicine 2014;127:124-131
Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia
SUMMARY: Normal B-cell activation and proliferation is dependent on B-cell receptor (BCR) signaling. This signaling is also important for initiation and progression of B-cell lymphoproliferative disorders. Bruton’s tyrosine kinase (BTK) is a member of the Tec family of kinases, downstream of the B-cell receptor and is predominantly expressed in B-cells. It is a mediator of B-cell receptor signaling in normal and transformed B-cells. Following binding of antigen to the BCR, Syk (Spleen Tyrosine Kinase), Lyn (member of the Src family of protein tyrosine kinases) and BTK (Bruton’s Tyrosine Kinase) are activated, with subsequent propagation through PI3K/Akt, MAPK, NF-κB pathways and resulting B-cell activation and proliferation. IMBRUVICA® (Ibrutinib, PCI-32765) is an oral, irreversible inhibitor of BTK and inhibits cell proliferation and promotes programmed cell death (Apoptosis).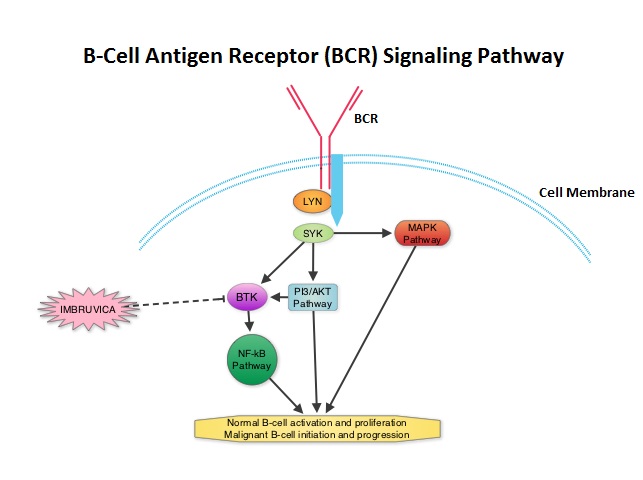 The FDA granted accelerated approval of IMBRUVICA® for the treatment of patients with Chronic Lymphocytic Leukemia (CLL) who had received at least one prior therapy. This approval was based on the outcomes in a select group of 48 patients who were a part of a larger group of 85 patients, enrolled in a multicenter single arm phase Ib/II trial. The median age was 67 years and 71% were male. Patients had a median number of 4 prior treatments and had an ECOG PS of 0-1. Patients in this group received IMBRUVICA® 420 mg PO daily until disease progression or unacceptable toxicity. The overall response rate was 58.3% as assessed by an independent review committee. No complete responses were seen and the response duration ranged from 5.6 to over 24 months. This analysis did not include data from those patients enrolled in the trial who received IMBRUVICA® 840 mg PO daily or those with Small Lymphocytic Lymphoma (N=37). The most common toxicities included fatigue, myalgias and arthralgias, cytopenias, nausea, diarrhea, fever and rash. Transient asymptomatic increase in lymphocyte count with resolution of lymphadenopathy and splenomegaly was common but resolved with continued treatment. The confirmatory RESONATE trial is a multicenter, randomized, open-label Phase III study in which single agent IMBRUVICA® was compared to single agent ARZERRA® (Ofatumumab) in patients with relapsed or refractory CLL or Small Lymphocytic Lymphoma . This was a part of the requirement by the FDA. Enrolled patients had measurable nodal disease and were not eligible for treatment with purine analog-based therapy. In this study, 391 patients who had received at least one prior therapy, were enrolled and randomized to receive 420 mg of IMBRUVICA® orally once daily or ARZERRA® given intravenously. Treatment was given over a period of 24 weeks until disease progression or unacceptable toxicity. Patients randomized to the ARZERRA® group on disease progression were allowed to receive treatment with IMBRUVICA®. The primary endpoint of this study was progression-free survival and the secondary endpoint was overall survival. Following recommendations from the Independent Data Monitoring Committee (IDMC), the study was stopped earlier, as the primary endpoint as well as an important secondary endpoint of the study were met. At the planned interim analysis, patients in the IMBRUVICA® group showed a statistically significant improvement in progression-free survival, the primary endpoint of the study as well as a statistically significant improvement in overall survival, the secondary endpoint of the trial. This data confirmed the efficacy of IMBRUVICA® and gives patients with CLL, an important new treatment option. Byrd JC, Furman RR, Coutre SE, et al. N Engl J Med 2013; 369:32-42
The FDA granted accelerated approval of IMBRUVICA® for the treatment of patients with Chronic Lymphocytic Leukemia (CLL) who had received at least one prior therapy. This approval was based on the outcomes in a select group of 48 patients who were a part of a larger group of 85 patients, enrolled in a multicenter single arm phase Ib/II trial. The median age was 67 years and 71% were male. Patients had a median number of 4 prior treatments and had an ECOG PS of 0-1. Patients in this group received IMBRUVICA® 420 mg PO daily until disease progression or unacceptable toxicity. The overall response rate was 58.3% as assessed by an independent review committee. No complete responses were seen and the response duration ranged from 5.6 to over 24 months. This analysis did not include data from those patients enrolled in the trial who received IMBRUVICA® 840 mg PO daily or those with Small Lymphocytic Lymphoma (N=37). The most common toxicities included fatigue, myalgias and arthralgias, cytopenias, nausea, diarrhea, fever and rash. Transient asymptomatic increase in lymphocyte count with resolution of lymphadenopathy and splenomegaly was common but resolved with continued treatment. The confirmatory RESONATE trial is a multicenter, randomized, open-label Phase III study in which single agent IMBRUVICA® was compared to single agent ARZERRA® (Ofatumumab) in patients with relapsed or refractory CLL or Small Lymphocytic Lymphoma . This was a part of the requirement by the FDA. Enrolled patients had measurable nodal disease and were not eligible for treatment with purine analog-based therapy. In this study, 391 patients who had received at least one prior therapy, were enrolled and randomized to receive 420 mg of IMBRUVICA® orally once daily or ARZERRA® given intravenously. Treatment was given over a period of 24 weeks until disease progression or unacceptable toxicity. Patients randomized to the ARZERRA® group on disease progression were allowed to receive treatment with IMBRUVICA®. The primary endpoint of this study was progression-free survival and the secondary endpoint was overall survival. Following recommendations from the Independent Data Monitoring Committee (IDMC), the study was stopped earlier, as the primary endpoint as well as an important secondary endpoint of the study were met. At the planned interim analysis, patients in the IMBRUVICA® group showed a statistically significant improvement in progression-free survival, the primary endpoint of the study as well as a statistically significant improvement in overall survival, the secondary endpoint of the trial. This data confirmed the efficacy of IMBRUVICA® and gives patients with CLL, an important new treatment option. Byrd JC, Furman RR, Coutre SE, et al. N Engl J Med 2013; 369:32-42
IMBRUVICA® (Ibrutinib)
The FDA on February 12, 2014 granted accelerated approval to IMBRUVICA® for the treatment of patients with Chronic Lymphocytic Leukemia (CLL) who have received at least one prior therapy. The FDA initially granted accelerated approval in November, 2013, for the treatment of patients with Mantle Cell Lymphoma (MCL) who have received at least one prior therapy. IMBRUVICA® is an oral capsule and is a product of Pharmacyclics, Inc.
Maintenance treatment with capecitabine and bevacizumab versus observation after induction treatment with chemotherapy and bevacizumab in metastatic colorectal cancer (mCRC) The phase III CAIRO3 study of the Dutch Colorectal Cancer Group (DCCG)
SUMMARY: Treatment of metastatic colorectal cancer with a combination of chemotherapy given along with AVASTIN® is well established. However the duration of therapy remains unclear and it is common to give drug holidays to patients. The outcome in patients who are given these drug holidays remains unclear. The CAIRO3 study is a phase III trial in which patients with previously untreated, unresectable metastatic colorectal cancer received induction treatment with six cycles of Capecitabine (XELODA®)/Oxaliplatin (ELOXATIN®) plus Bevacizumab (AVASTIN®) – CAPOX-B. Patients who had not progressed during induction and had reponses or had stable disease (N=558) were then randomized to receive either XELODA® at 625 mg/m2 twice daily along with AVASTIN® at 7.5 mg/kg every 3 weeks or be observed. Upon first progression, patients in both treatment groups were treated with CAPOX-B until second progression and this was considered the primary endpoint for this study. Secondary endpoints included Overall Survival (OS). Median follow up was 40 months. The median time to second progression from randomization was 19.8 months in the maintenance group and 15 months in the observation group (HR=0.63; P<0.001) The time to first progression in the maintenance treatment group was 8.5 months versus 4.1 months in the observation group (HR 0.41; P<0.001). The time to second progression following treatment with CAPOX-B was 11.8 months in the maintenance group versus 10.5 months for the observation group (HR 0.77; P=0.007), representing a 23% reduction in the risk of progression. The adjusted median OS was 21.7 months with maintenance treatment and 18.2 months in the observation group (HR=0.80; P=0.035). Treatment was well tolerated with slight increase in hand-foot syndrome and neurotoxicity in the maintenance group. Based on this data, the authors recommended maintenance treatment with XELODA® and AVASTIN® until progression or unacceptable toxicity, following 6 cycles of efficacious treatment with CAPOX-B. It is important to note that in the SAKK 41/06 trial conducted by the Swiss Group, observation alone was non-inferior to single agent maintenance AVASTIN® following initial chemotherapy, suggesting that the addition of fluoropyrimidine (XELODA®) chemotherapy to AVASTIN® as maintenance treatment, improves time to progression and median OS in patients with metastatic colorectal cancer. Koopman M, Simkens LH, Ten Tije, AJ et al. J Clin Oncol 31, 2013 (suppl; abstr 3502)
Clinical Cancer Advances 2013 Annual Report on Progress Against Cancer From the American Society of Clinical Oncology
SUMMARY: Immune checkpoints are cell surface inhibitory proteins/receptors that harness the immune system and prevent uncontrolled immune reactions. Immune checkpoints are an area of increasing interest as they utilize the patient’s immune system to reject cancer cells. Survival of cancer cells in the human body may be to a significant extent, related to their ability to escape immune surveillance, by inhibiting T lymphocyte Activation .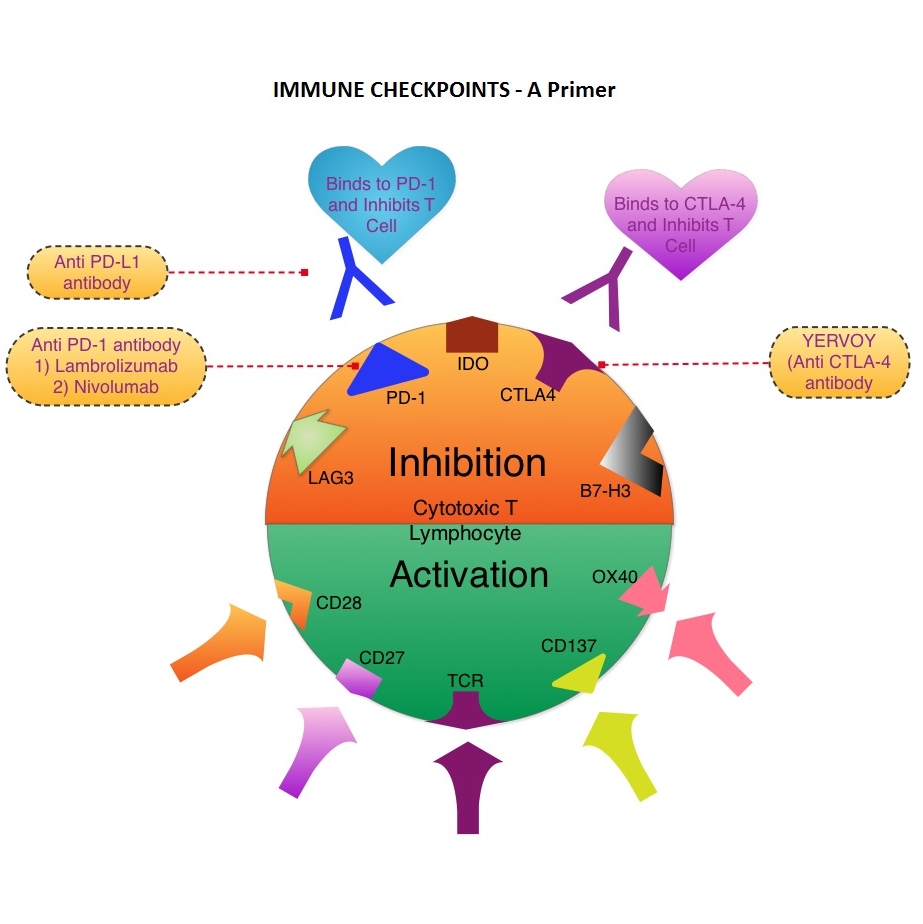 The T cells of the immune system play a very important role in modulating the immune system. EFFECTOR T cells include Cytotoxic T cells, Helper T cells, and Natural Killer (NK) cells, that enable the immune system to destroy cancer cells and pathogens. The REGULATORY T cells however, suppress immune response. Under normal circumstances, inhibition of an intense immune response and switching off the EFFECTOR T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. The mechanism can be compared to a lock and key where the appropriate Ligand (KEY) binds to the Immune checkpoint protein/receptor (LOCK) and activates or inhibits a T lymphocyte. With the ongoing understanding of tumor immunology and the recognition of Immune checkpoint proteins, researchers have focused on the development of antibodies that either target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4, PD-1, IDO, etc. (LOCK) or target the inhibitory soluble Ligands or antigens that are located on the surface of certain cancer cells (KEY) that bind to these Immune check point proteins/receptors. By doing so, one would expect to unleash the EFFECTOR T cells resulting in T cell proliferation, activation and a therapeutic response. The first immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab), an antibody that blocks Immune checkpoint protein/receptor CTLA-4, was approved by the FDA in March 2011 and has been shown to prolong overall survival in patients with previously treated unresectable or metastatic melanoma. The next immune check point protein/receptor studied for targeted therapy was PD-1. Lambrolizumab (MK-3475) is a humanized anti–PD-1 monoclonal antibody that demonstrated a 38% rapid and durable response rate and a more than 7 month median progression-free survival in patients with advanced melanoma, regardless of their prior therapy with YERVOY®. Nivolumab, another PD-1 targeted antibody demonstrated remarkable efficacy in a Phase I study with an overall response rate of 30%, median survival of 16.8 months and a 2 year survival of 44%. Based on this provocative data, a combination of Nivolumab and YERVOY® were studied in patients with advanced Stage III or IV melanoma who had received up to three prior therapies.. The idea was to block both the Immune checkpoints, PD-1 and CTLA-4, for improved efficacy. Fifty three (N=53) patients were treated with a combination of these two agents and 33 patients received these agents sequentially. Indeed, the highest response rate was over 50% in the combination group with 30% of these patients experienced a more than 80% response rate at 12 weeks of treatment whereas the response rate in the sequential treatment group was 20%. This preliminary study confirmed that blocking multiple Immune checkpoint proteins/receptors may result in rapid and durable responses in patients with advanced malignant melanoma. Phase III studies are underway to confirm this efficacy data and this concept is also being studied in other tumor types. Targeting/inhibiting the ligands (KEY) and preventing their binding to the Immune checkpoint protein/receptor, is another approach to stimulate antitumor immune response. PD-L1 protein (Ligand) which is often elevated in melanoma tumor cells, bind to PD-1 check point protein/receptor and can inhibit T cells and escape immune surveillance. An investigational PD-L1 targeted (Ligand targeted) engineered antibody (MPDL3280A) demonstrated a rapid response in 26% of the 45 patients with metastatic melanoma and the benefit was more so in those tumors expressing PD-L1. Promising activity has also been seen in advanced renal cell carcinoma. Antibodies targeting the Immune checkpoint receptor/protein or the Ligands binding to these receptors, are being developed, to carry payloads that are lethal to the checkpoint protein/receptor or Ligand. In conclusion, identifying as well as inhibiting certain Immune checkpoint proteins/receptors and/or Ligands that bind to these receptors, may give us new insights in the field of tumor immunology, resulting in better outcome for our cancer patients. Patel JD, Krilov L, Adams S, et al. J Clin Oncol 2013;32:129-160
The T cells of the immune system play a very important role in modulating the immune system. EFFECTOR T cells include Cytotoxic T cells, Helper T cells, and Natural Killer (NK) cells, that enable the immune system to destroy cancer cells and pathogens. The REGULATORY T cells however, suppress immune response. Under normal circumstances, inhibition of an intense immune response and switching off the EFFECTOR T cells of the immune system, is an evolutionary mechanism and is accomplished by Immune checkpoints or gate keepers. The mechanism can be compared to a lock and key where the appropriate Ligand (KEY) binds to the Immune checkpoint protein/receptor (LOCK) and activates or inhibits a T lymphocyte. With the ongoing understanding of tumor immunology and the recognition of Immune checkpoint proteins, researchers have focused on the development of antibodies that either target the membrane bound inhibitory Immune checkpoint proteins/receptors such as CTLA-4, PD-1, IDO, etc. (LOCK) or target the inhibitory soluble Ligands or antigens that are located on the surface of certain cancer cells (KEY) that bind to these Immune check point proteins/receptors. By doing so, one would expect to unleash the EFFECTOR T cells resulting in T cell proliferation, activation and a therapeutic response. The first immune checkpoint protein to be clinically targeted was CTLA-4. YERVOY® (Ipilimumab), an antibody that blocks Immune checkpoint protein/receptor CTLA-4, was approved by the FDA in March 2011 and has been shown to prolong overall survival in patients with previously treated unresectable or metastatic melanoma. The next immune check point protein/receptor studied for targeted therapy was PD-1. Lambrolizumab (MK-3475) is a humanized anti–PD-1 monoclonal antibody that demonstrated a 38% rapid and durable response rate and a more than 7 month median progression-free survival in patients with advanced melanoma, regardless of their prior therapy with YERVOY®. Nivolumab, another PD-1 targeted antibody demonstrated remarkable efficacy in a Phase I study with an overall response rate of 30%, median survival of 16.8 months and a 2 year survival of 44%. Based on this provocative data, a combination of Nivolumab and YERVOY® were studied in patients with advanced Stage III or IV melanoma who had received up to three prior therapies.. The idea was to block both the Immune checkpoints, PD-1 and CTLA-4, for improved efficacy. Fifty three (N=53) patients were treated with a combination of these two agents and 33 patients received these agents sequentially. Indeed, the highest response rate was over 50% in the combination group with 30% of these patients experienced a more than 80% response rate at 12 weeks of treatment whereas the response rate in the sequential treatment group was 20%. This preliminary study confirmed that blocking multiple Immune checkpoint proteins/receptors may result in rapid and durable responses in patients with advanced malignant melanoma. Phase III studies are underway to confirm this efficacy data and this concept is also being studied in other tumor types. Targeting/inhibiting the ligands (KEY) and preventing their binding to the Immune checkpoint protein/receptor, is another approach to stimulate antitumor immune response. PD-L1 protein (Ligand) which is often elevated in melanoma tumor cells, bind to PD-1 check point protein/receptor and can inhibit T cells and escape immune surveillance. An investigational PD-L1 targeted (Ligand targeted) engineered antibody (MPDL3280A) demonstrated a rapid response in 26% of the 45 patients with metastatic melanoma and the benefit was more so in those tumors expressing PD-L1. Promising activity has also been seen in advanced renal cell carcinoma. Antibodies targeting the Immune checkpoint receptor/protein or the Ligands binding to these receptors, are being developed, to carry payloads that are lethal to the checkpoint protein/receptor or Ligand. In conclusion, identifying as well as inhibiting certain Immune checkpoint proteins/receptors and/or Ligands that bind to these receptors, may give us new insights in the field of tumor immunology, resulting in better outcome for our cancer patients. Patel JD, Krilov L, Adams S, et al. J Clin Oncol 2013;32:129-160
Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer The phase III DECISION trial
SUMMARY: Over 90% of all Thyroid cancers are classified as Differentiated Thyroid Cancers (DTC) with Papillary, Follicular and Hürthle cell histologies. Approximately 5% to 15% of these patients develop resistance to RadioActive Iodine (RAI). NEXAVAR® is a multi-targeted tyrosine kinase inhibitor and prevents cancer growth by inhibiting multiple kinases that are involved in cell proliferation and angiogenesis. These kinases include Raf, VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-B, KIT, FLT-3 and RET.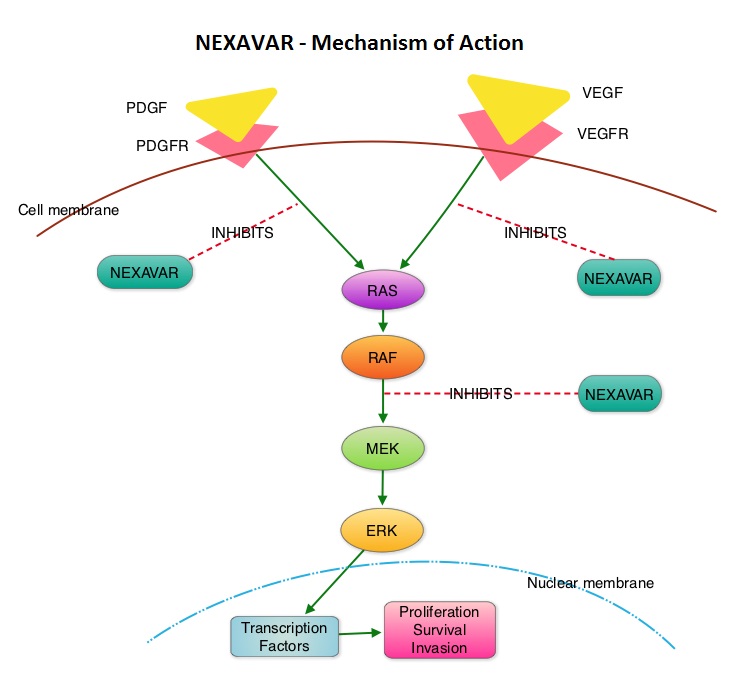 The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)