
Written by: Thomas E Boyd, MD, Texas Oncology
Content Sponsored by: Bristol-Myers Squibb
Dr. Boyd is a paid consultant for BMS and was compensated for his contribution in drafting this article.
Acute myeloid leukemia (AML) is a deadly disease that is more common in older adults.1 In 2021, it is estimated that there will be 20,240 new cases of AML in the United States, representing 1.1% of all new cancer cases.1 Additionally, there will be an estimated 11,400 deaths due to AML, representing 1.9% of all cancer deaths in the US in 2021.1 Once a patient is diagnosed with AML, beginning treatment as soon as possible is essential for disease management and survival.2
Currently, patients usually follow one of two paths for initial treatment of AML: conventional intensive induction chemotherapy or a less intensive option, with some patients going onto hematopoietic stem cell transplant after either initial treatment.3 The choice of treatment path is based on both patient- and disease-related characteristics such as medical fitness, age, cytogenetic and molecular testing, and risk of adverse events.2 With the progress seen in our understanding of the biology of AML, our knowledge of the molecular underpinnings of AML pathology has greatly improved over the years.4 This deeper understanding of disease at the molecular level has helped pave the way for a wave of approved therapies, with several new drug approvals beginning in 2017.5
A major goal of AML treatment is achieving remission.6 However, proliferative AML cells may still persist in remission, leading to a risk of relapse.4 Continued treatment of AML in first remission may improve overall survival.8
The large, multicenter QUAZAR®AML-001 trial established the efficacy and safety of ONUREG®(azacitidine) tablets, the first and only FDA-approved continued AML treatment for patients in first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy.8 Eligible patients were ages 55 years or older, diagnosed with AML, were within 4 months of achieving first CR or CRi with intensive induction chemotherapy, and may have received consolidation therapy.8 Patients could not enroll in the study if they were candidates for hematopoietic stem cell transplantation at the time of screening.8 Additional criteria included an ECOG performance status (PS) 0-3 and intermediate- or poor-risk cytogenetics, defined as normal cytogenetics +8, t(9;11), or other undefined, and complex (≥3 abnormalities): -5; 5q-; -7; 7q-; 11q23 – non t(9;11); inv(3); t(3;3); t(6;9); or t(9;22), respectively.8
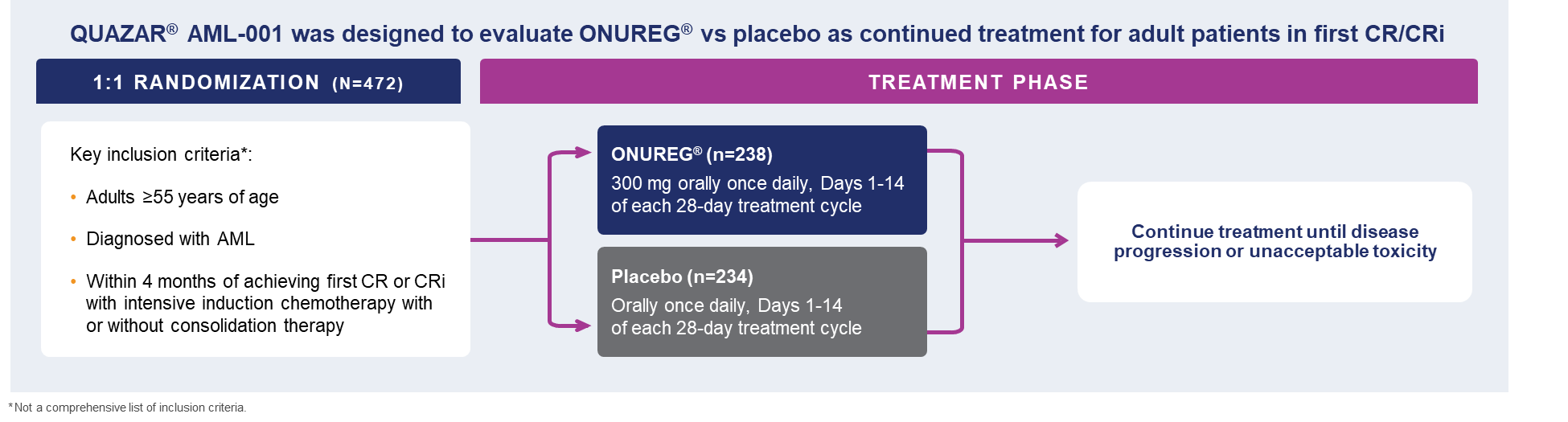 A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8
A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8
With a >2-year median overall survival and a statistically significant survival benefit of ~10 months for patients with AML in first remission compared to placebo, ONUREG met its primary endpoint (24.7 months in the treated arm vs 14.8 months in the placebo arm, hazard ratio (HR) 0.69, 95% confidence interval (CI): 0.55-0.86; P=0.0009).8
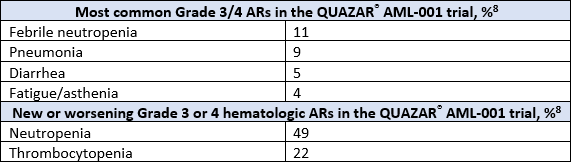
The most common adverse reactions (ARs, ≥ 10%) associated with ONUREG® treatment included nausea, vomiting, diarrhea, fatigue/asthenia, constipation, pneumonia, abdominal pain, arthralgia, decreased appetite, febrile neutropenia, dizziness, and pain in extremity.8 Serious ARs occurred in 15% of patients who received ONUREG®, with select Grade 3/4 ARs shown in the table below.8 Eight percent of patients permanently discontinued ONUREG®, 35% of patients required a treatment interruption due to an AR, and 14% of patients required a dose reduction.8
ONUREG® is approved for continued treatment of adult patients with acute myeloid leukemia who achieved first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy.8 ONUREG® is an oral hypomethylating agent, offering a convenient, once-daily dosing that patients can take at home.8 However, it is important to emphasize that ONUREG® should not be substituted for intravenous or subcutaneous azacitidine, because the indications and dosing regimen differ between these formulations.8 As the first and only FDA-approved continued AML treatment for patients in first remission, ONUREG® remains an option for appropriate patients.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
ONUREG® is contraindicated in patients with known severe hypersensitivity to azacitidine or its components.
WARNINGS AND PRECAUTIONS
Risks of Substitution with Other Azacitidine Products
Due to substantial differences in the pharmacokinetic parameters, the recommended dose and schedule for ONUREG® are different from those for the intravenous or subcutaneous azacitidine products. Treatment of patients using intravenous or subcutaneous azacitidine at the recommended dosage of ONUREG® may result in a fatal adverse reaction. Treatment with ONUREG® at the doses recommended for intravenous or subcutaneous azacitidine may not be effective. Do not substitute ONUREG® for intravenous or subcutaneous azacitidine.
Myelosuppression
New or worsening Grade 3 or 4 neutropenia and thrombocytopenia occurred in 49% and 22% of patients who received ONUREG®. Febrile neutropenia occurred in 12%. A dose reduction was required for 7% and 2% of patients due to neutropenia and thrombocytopenia. Less than 1% of patients discontinued ONUREG® due to either neutropenia or thrombocytopenia. Monitor complete blood counts and modify the dosage as recommended. Provide standard supportive care, including hematopoietic growth factors, if myelosuppression occurs.
Increased Early Mortality in Patients with Myelodysplastic Syndromes (MDS)
In AZA-MDS-003, 216 patients with red blood cell transfusion-dependent anemia and thrombocytopenia due to MDS were randomized to ONUREG® or placebo. 107 received a median of 5 cycles of ONUREG® 300 mg daily for 21 days of a 28-day cycle. Enrollment was discontinued early due to a higher incidence of early fatal and/or serious adverse reactions in the ONUREG® arm compared with placebo. The most frequent fatal adverse reaction was sepsis. Safety and effectiveness of ONUREG® for MDS have not been established. Treatment of MDS with ONUREG® is not recommended outside of controlled trials.
Embryo-Fetal Toxicity
ONUREG® can cause fetal harm when administered to a pregnant woman. Azacitidine caused fetal death and anomalies in pregnant rats via a single intraperitoneal dose less than the recommended human daily dose of oral azacitidine on a mg/m2 basis. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ONUREG® and for at least 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with ONUREG® and for at least 3 months after the last dose.
ADVERSE REACTIONS
Serious adverse reactions occurred in 15% of patients who received ONUREG®. Serious adverse reactions in ≥2% included pneumonia (8%) and febrile neutropenia (7%). One fatal adverse reaction (sepsis) occurred in a patient who received ONUREG®.
Most common (≥10%) adverse reactions with ONUREG® vs placebo were nausea (65%, 24%), vomiting (60%, 10%), diarrhea (50%, 21%), fatigue/asthenia (44%, 25%), constipation (39%, 24%), pneumonia (27%, 17%), abdominal pain (22%, 13%), arthralgia (14%, 10%), decreased appetite (13%, 6%), febrile neutropenia (12%, 8%), dizziness (11%, 9%), pain in extremity (11%, 5%).
LACTATION
There are no data regarding the presence of azacitidine in human milk or the effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with ONUREG® and for 1 week after the last dose.
Please see full Prescribing Information for ONUREG®.
References
1. National Cancer Institute. SEER Cancer Statistics Factsheets: Acute Myeloid Leukemia. http://seer.cancer.gov/statfacts/html/amyl.html. Accessed April 21, 2021.
2. Medeiros BC, Satram S. Real world treatment patterns and comparative effectiveness among elderly patients with acute myeloid leukemia in the United States. Ann Hematol Oncol. 2020;7(1):1283.
3. Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29(5):487-494.
4. Brinda B, Khan I, Parkin B, Konig H. The rocky road to personalized medicine in acute myeloid leukaemia. J Cell Mol Med. 2018;22(3):1411-1427.
5. Lai C, Doucette K, Norsworthy K. Recent drug approvals for acute myeloid leukemia. J Hematol Oncol. 2019;12(100):1-20.
6. Medeiros BC. Interpretation of clinical endpoints in trials of acute myeloid leukemia. Leuk Res. 2018;68:32-29.
7. Medeiros BC, Chan SM, Daver NG, Jonas BA, Pollyea DA. Optimizing survival outcomes with post-remission therapy in acute myeloid leukemia. Am J Hematol. 2019;94:803-811.
8. ONUREG® [Prescribing Information]. Summit, NJ: Celgene Corporation; 2021.
© 2021 Celgene Corporation.
ONUREG is a trademark of Celgene Corporation, a Bristol-Myers Squibb company.
QUAZAR is a registered trademark of Celgene Corporation, a Bristol-Myers Squibb company.
1/22 2011-US-2100199
