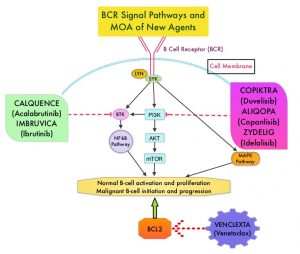
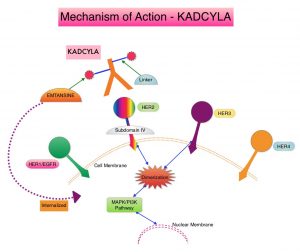
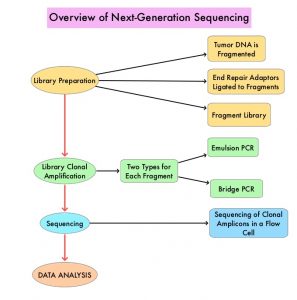
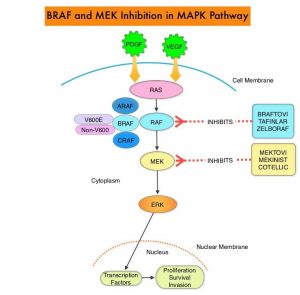
SUMMARY: The American Cancer Society estimates that in 2020, there will be an estimated 1.8 million new cancer cases diagnosed and 606,520 cancer deaths in the United States. Immunotherapy with Immune Checkpoint Inhibitors (ICIs) has revolutionized cancer care and has become one of the most effective treatment options by improving Overall Response Rate and prolongation of survival across multiple tumor types. These agents target Programmed cell Death protein-1 (PD-1), Programmed cell Death Ligand-1 (PD-L1), Cytotoxic T-Lymphocyte-Associated protein-4 (CTLA-4), and many other important regulators of the immune system. Biomarkers predicting responses to ICI’s include Tumor Mutational Burden (TMB), Mismatch Repair (MMR) status, and Programmed cell Death Ligand 1 (PD‐L1) expression. Other biomarkers such as Tumor Infiltrating Lymphocytes (TILs), TIL‐derived Interferon‐γ, Neutrophil‐to‐Lymphocyte ratio, and peripheral cytokines, have also been proposed as predictors of response.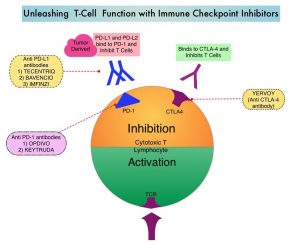
Immune Checkpoint Inhibitors enhance antitumor immunity by unleashing the T cells. However, this benefit may vary among patients and tumor types. Sex is a biological variable that affects immune responses. Women tend to mount stronger innate as well as adaptive immune responses, than men. (Innate immunity is inherently present in the body, whereas Adaptive immunity occurs in response to exposure to a foreign substance). This can translate into greater efficacy with vaccines and more rapid clearance of pathogens. Aging alters immune responses and adaptive immunity becomes less functional. Altered ECOG Performance Status has also been associated with poor immune response. Several other studies have been published looking at these variables, with conflicting results.
To address these discordant results, the authors performed a meta-analysis to examine the potential association of sex, age, and ECOG PS with immunotherapy survival benefit in patients with advanced cancer. This study was limited to randomized clinical trials that compared Overall Survival (OS) in patients with advanced cancer treated with ICI immunotherapy versus non-ICI control therapy. Data sources such as PubMed, Web of Science, Embase, and Scopus were searched and a total of 37 Phase II or III randomized clinical trials involving 23,760 patients were included in the analysis. Of these, 13 trials evaluated ICIs as first-line therapy. The most common cancer types studied were Non-Small Cell Lung Cancer (N= 14). The most common immune checkpoint inhibitors used were PD-1/PD-L1 inhibitors (N=25). The main Outcomes and Measures were the difference in survival benefit of ICIs between sex, age (less than 65 versus 65 years or more), ECOG PS (0 versus 1 or more), as well as the outcomes stratified by cancer type, line of therapy, agent of immunotherapy, and immunotherapy strategy (ICI alone or ICI combined with non-ICI) in the intervention arm.
The authors noted that Overall Survival benefit with ICI immunotherapy treatment was found for both men (HR=0.75) and women (HR=0.79), for both younger and less than 65 years (HR=0.77) and 65 years or older (HR=0.78) patients, and for both, patients with ECOG Performance Status 0 (HR=0.81) and PS greater than or equal to 1 (HR=0.79). There was no significant difference of relative benefit from immunotherapy over control therapy in patients of different sex (P=0 .25), age (P=0.94), or ECOG PS (P=0.74). Further, there was no significant difference found in subgroup analyses by cancer type, line of therapy, agent of immunotherapy, and immunotherapy strategy in the intervention arm.
It was concluded that the results of this meta-analysis suggest that immunotherapy with ICIs may confer a survival benefit in the treatment of advanced cancer, regardless of patient sex, age, and performance status, and should not be restricted based on these characteristics.
Association of Sex, Age, and Eastern Cooperative Oncology Group Performance Status With Survival Benefit of Cancer Immunotherapy in Randomized Clinical Trials. A Systematic Review and Meta-analysis. Yang F, Markovic SN, Molina JR, et al. JAMA Netw Open. 2020;3(8):e2012534. doi:10.1001/jamanetworkopen.2020.12534