SUMMARY: Over 90% of all Thyroid cancers are classified as Differentiated Thyroid Cancers (DTC) with Papillary, Follicular and Hürthle cell histologies. Approximately 5% to 15% of these patients develop resistance to RadioActive Iodine (RAI). NEXAVAR® is a multi-targeted tyrosine kinase inhibitor and prevents cancer growth by inhibiting multiple kinases that are involved in cell proliferation and angiogenesis. These kinases include Raf, VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-B, KIT, FLT-3 and RET.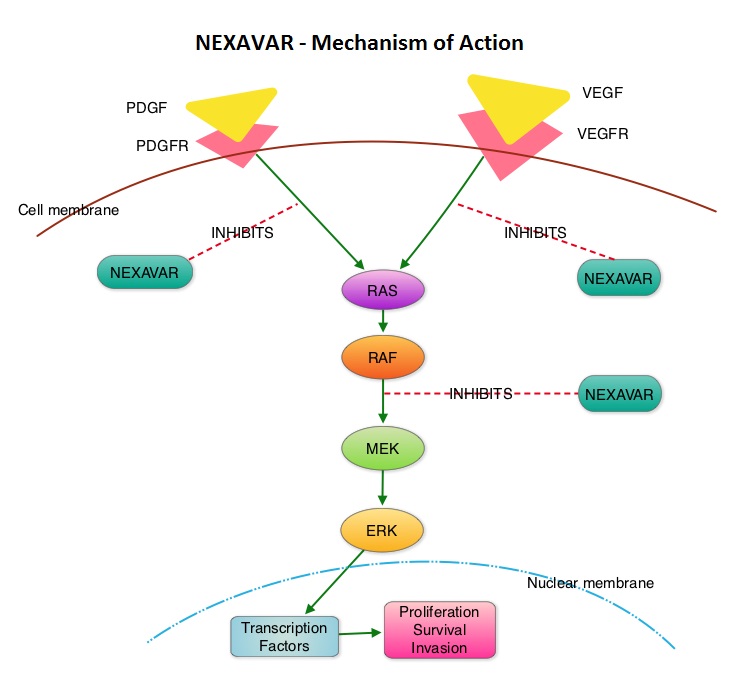 The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
Category: Hem/Onc Updates
Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer The phase III DECISION trial
SUMMARY: Over 90% of all Thyroid cancers are classified as Differentiated Thyroid Cancers (DTC) with Papillary, Follicular and Hürthle cell histologies. Approximately 5% to 15% of these patients develop resistance to RadioActive Iodine (RAI). NEXAVAR® is a multi-targeted tyrosine kinase inhibitor and prevents cancer growth by inhibiting multiple kinases that are involved in cell proliferation and angiogenesis. These kinases include Raf, VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-B, KIT, FLT-3 and RET.  The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this may impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this may impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
Enzalutamide in men with chemotherapy-naive metastatic prostate cancer (mCRPC) Results of phase III PREVAIL study
SUMMARY: Prostate Cancer is driven by androgens (primarily testosterone) and androgen signaling pathways. There is evidence to suggest that prostate cancer cells continue to depend on androgen receptor (AR) signaling even in an androgen-deprived environment. Therefore, targeting AR and AR signaling pathways remains a rational approach in the treatment of Castration Resistant Prostate Cancer (CRPC). The first generation anti-androgen agents such as EULEXIN® (Flutamide), CASODEX® (Bicalutamide) and NILANDRON® (Nilutamide) act by binding to the Androgen Receptor (AR) and prevent the activation of the AR and subsequent up-regulation of androgen responsive genes. They may also accelerate the degradation of the AR. These agents have a range of pharmacologic activity from being pure anti-androgens to androgen agonists.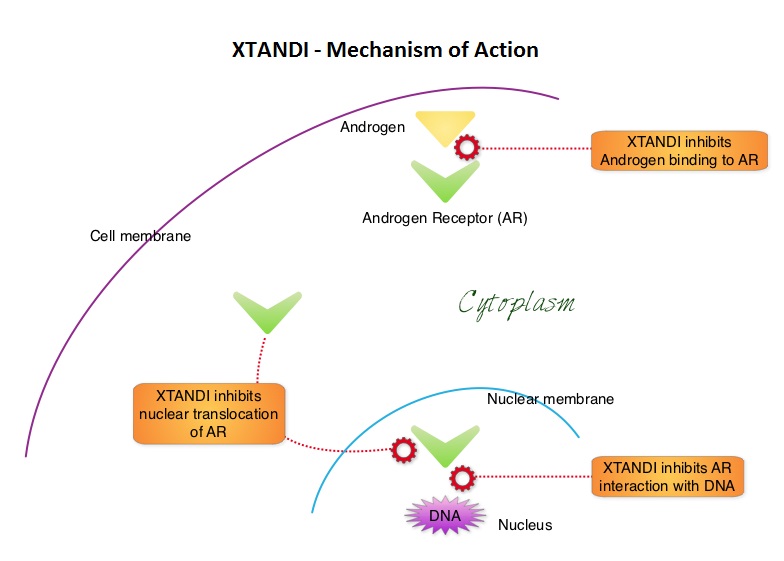 XTANDI® (Enzalutamide) is a second-generation anti-androgen with no reported agonistic effects. It competitively inhibits androgens and AR binding to androgens as well as AR nuclear translocation and interaction with DNA. It thus inhibits several steps in the AR signaling pathway. XTANDI® was first approved by the FDA in 2012, for the treatment of patients with metastatic CRPC who have previously received TAXOTERE® (Docetaxel) based chemotherapy. The PREVAIL study is a double-blind, placebo-controlled, phase III trial in which 1,717 chemotherapy-naive patients with mCRPC (metastatic Castrate Resistant Prostate Cancer) were randomly assigned 1:1 to receive either XTANDI® 160 mg/day or placebo. Prior treatment with surgery or radiation therapy for their primary tumor, as well as hormonal intervention with a LHRH (Luteinizing Hormone Releasing Hormone) agonist or first-generation anti-androgen was allowed. The two co-primary endpoints were Overall Survival (OS) and radiographic Progression Free Survival (rPFS), as measured by bone scans and CT scans. At the time of preplanned interim analysis, XTANDI® demonstrated a statistically significant benefit over placebo with a 30% reduction in risk of death (OS: HR= 0.70; P< 0.0001) and an 81% reduction in risk of radiographic Progression Free Survival (rPFS: HR 0.19; P< 0.0001). Further, the response rates were meaningful with 20% complete responses and 39% partial responses (59% Response Rate) compared with 5% Response Rate in the placebo group (P<0.0001). XTANDI® also significantly delayed the median time to chemotherapy by 17 months compared with those who took placebo (P<0.0001). Based on the results of this interim analysis, the Independent Data Monitoring Committee recommended stopping the study and allowing patients in the placebo group to receive XTANDI®. XTANDI® was well tolerated and the most common side effects were hot flashes, weight gain, fatigue, constipation, back and joint pain. The authors concluded that XTANDI® significantly improves OS and rPFS in patients with chemotherapy-naive mCRPC and can significantly delay the need for chemotherapeutic intervention. Beer TM, Armstrong AJ, Sternberg CN, et al. J Clin Oncol 32, 2014 (suppl 4; abstr LBA1)
XTANDI® (Enzalutamide) is a second-generation anti-androgen with no reported agonistic effects. It competitively inhibits androgens and AR binding to androgens as well as AR nuclear translocation and interaction with DNA. It thus inhibits several steps in the AR signaling pathway. XTANDI® was first approved by the FDA in 2012, for the treatment of patients with metastatic CRPC who have previously received TAXOTERE® (Docetaxel) based chemotherapy. The PREVAIL study is a double-blind, placebo-controlled, phase III trial in which 1,717 chemotherapy-naive patients with mCRPC (metastatic Castrate Resistant Prostate Cancer) were randomly assigned 1:1 to receive either XTANDI® 160 mg/day or placebo. Prior treatment with surgery or radiation therapy for their primary tumor, as well as hormonal intervention with a LHRH (Luteinizing Hormone Releasing Hormone) agonist or first-generation anti-androgen was allowed. The two co-primary endpoints were Overall Survival (OS) and radiographic Progression Free Survival (rPFS), as measured by bone scans and CT scans. At the time of preplanned interim analysis, XTANDI® demonstrated a statistically significant benefit over placebo with a 30% reduction in risk of death (OS: HR= 0.70; P< 0.0001) and an 81% reduction in risk of radiographic Progression Free Survival (rPFS: HR 0.19; P< 0.0001). Further, the response rates were meaningful with 20% complete responses and 39% partial responses (59% Response Rate) compared with 5% Response Rate in the placebo group (P<0.0001). XTANDI® also significantly delayed the median time to chemotherapy by 17 months compared with those who took placebo (P<0.0001). Based on the results of this interim analysis, the Independent Data Monitoring Committee recommended stopping the study and allowing patients in the placebo group to receive XTANDI®. XTANDI® was well tolerated and the most common side effects were hot flashes, weight gain, fatigue, constipation, back and joint pain. The authors concluded that XTANDI® significantly improves OS and rPFS in patients with chemotherapy-naive mCRPC and can significantly delay the need for chemotherapeutic intervention. Beer TM, Armstrong AJ, Sternberg CN, et al. J Clin Oncol 32, 2014 (suppl 4; abstr LBA1)
Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II) an international, double-blind, randomised placebo-controlled trial
SUMMARY: NOLVADEX® (Tamoxifen) is a Selective Estrogen Receptor Modulator (SERM), approved by the FDA to also reduce the incidence of breast cancer in women considered to be at high risk (ChemoPrevention). This agent however has been linked to endometrial cancer and thromboembolic phenomenon in some women. ARIMIDEX® is a non-steroidal Aromatase Inhibitor proven to be more efficacious than NOLVADEX® both in metastatic and adjuvant settings. Similar to NOLVADEX®, ARIMIDEX® also prevents the occurrence of new primary tumors in the contralateral breast, in postmenopausal females. With this background, the International Breast Cancer Intervention Study (IBIS) -II trial enrolled 3864 postmenopausal women considered to be at increased risk of breast cancer and randomized them to receive either ARIMIDEX® (Anastrazole) 1 mg QD (N=1920) or Placebo QD (N=1944)for 5 years. Patients were considered to be at high risk if they had a family history of breast cancer, atypical ductal hyperplasia, lobular carcinoma in-situ or dense breast tissue. The median age was 59 years. The primary end point was histologically confirmed Invasive breast cancer or Ductal Carcinoma In- Situ. At a median follow up of 5 years, the incidence of breast cancer in the ARIMIDEX® group was 2% and in the placebo group was 4%. The predicted cumulative incidence of breast cancer after 7 years was 2.8% in the ARIMIDEX® group and 5.6% in the placebo group (HR=0.47; P<0.0001). This represented a 53% reduction in the risk of breast cancer. The adverse events were comparable in both groups with slight increase in the musculoskeletal and vasomotor events noted in the ARIMIDEX® group. The authors concluded that ARIMIDEX® reduced the risk of primary breast cancer by more than 50% in high risk postmenopausal women. Other unrelated studies have shown that acceptance of breast cancer prevention with medications (ChemoPrevention) appeared to be related to education and income, putting emphasis on education and adequate counseling of women, considered to be at high risk. Cuzick J, Sestak I, Forbes JF, et al. The Lancet, Early Online Publication, 12 December 2013
Ruxolitinib for Myelofibrosis – An Update of Its Clinical Effects
SUMMARY: MyeloFibrosis (MF) is a MyeloProliferative Neoplasm (MPN) characterized by a ineffective hematopoiesis, progressive fibrosis of the bone marrow and potential for leukemic transformation. This stem cell disorder is Philadelphia Chromosome negative and manifestations include anemia, splenomegaly and related symptoms such as abdominal distension and discomfort with early satiety. Cytokine driven debilitating symptoms such as fatigue, fever, night sweats, weight loss, pruritus and bone or muscle pain can further impact an individual’s quality of life. Myelofibrosis can be primary (PMF) or secondary to Polycythemia Vera (PV) or Essential Thrombocythemia (ET). The JAK-STAT signaling pathway has been implicated in the pathogenesis of Myelofibrosis. 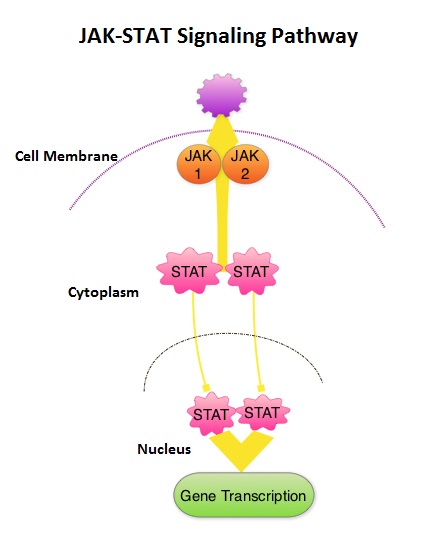 This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling. JAKAFI® is a potent JAK1 and JAK2 inhibitor and exerts its mechanism of action by targeting and inhibiting the dysregulated JAK2-STAT signaling pathway. The FDA approval of JAKAFI® for the treatment of Intermediate and high risk Myelofibrosis was based on 2 phase III trials – COMFORT (Controlled Myelofibrosis Study with Oral JAK1/JAK2 Inhibitor Treatment) – I and COMFORT-II studies. In COMFORT-I study, 309 intermediate or high risk patients were randomized to receive either JAKAFI® (N=155) or Placebo (N=154). The primary end point of a 35% or more reduction in spleen size at 24 weeks was noted in 42% of those who received JAKAFI® vs 0.7% in the placebo group (P<0.0001). Most patients in the JAKAFI® group had some reduction in the spleen volume whereas majority of those in the placebo arm had increase in splenomegaly. There was a 46% reduction in the TSS (Total Symptom Score) at week 24 in the JAKAFI® group compared to 5% in the placebo group and majority of patients in the later group had worsening of symptoms (P<0.0001). When JAKAFI® was compared to Best Available Therapy (BAT) in the COMFORT-II study, 28% of the patients in the JAKAFI® group met the primary endpoint of a 35% or more reduction in the spleen volume at 48 weeks compared to none in the BAT group (P<0.0001). Over 55% had a mean decrease in spleen size in the JAKAFI® compared to a 4% mean increase in the BAT group. The 2 year follow up analyses from both these trials showed improved overall survival and a reduction in the risk of death for patients randomized to JAKAFI®, compared to those in the control groups. There was weight gain with alleviation of cachexia and improvements in splenomegaly and symptoms were durable. This benefit was seen in patients regardless of JAK mutations. It remains to be seen if JAKAFI® will benefit patients with Polycythemia Vera and Essential Thrombocythemia. Kantarjian HM, Silver RT, Komrokji RS, et al. Clinical Lymphoma Myeloma and Leukemia 2013; 13:638-645
This pathway normally is responsible for passing information from outside the cell through the cell membrane to the DNA in the nucleus for gene transcription. Janus Kinase (JAK) family of tyrosine kinases are cytoplasmic proteins and include JAK1, JAK2, JAK3 and TYK2. JAK1 helps propagate the signaling of inflammatory cytokines whereas JAK2 is essential for growth and differentiation of hematopoietic stem cells. These tyrosine kinases mediate cell signaling by recruiting STAT’s (Signal Transducer and Activator of Transcription), with resulting modulation of gene expression. In patients with MPN, the aberrant myeloproliferation is the result of dysregulated JAK2-STAT signaling as well as excess production of inflammatory cytokines associated with this abnormal signaling. These cytokines contribute to the symptoms often reported by patients with MF. JAK2 mutations such as JAK2 V617F are seen in approximately 60% of the patients with PMF and ET and 95% of patients with PV. Unlike CML where the BCR-ABL fusion gene triggers the disease, JAK2 mutations are not initiators of the disease and are not specific for MPN. Further, several other genetic events may contribute to the abnormal JAK2-STAT signaling. JAKAFI® is a potent JAK1 and JAK2 inhibitor and exerts its mechanism of action by targeting and inhibiting the dysregulated JAK2-STAT signaling pathway. The FDA approval of JAKAFI® for the treatment of Intermediate and high risk Myelofibrosis was based on 2 phase III trials – COMFORT (Controlled Myelofibrosis Study with Oral JAK1/JAK2 Inhibitor Treatment) – I and COMFORT-II studies. In COMFORT-I study, 309 intermediate or high risk patients were randomized to receive either JAKAFI® (N=155) or Placebo (N=154). The primary end point of a 35% or more reduction in spleen size at 24 weeks was noted in 42% of those who received JAKAFI® vs 0.7% in the placebo group (P<0.0001). Most patients in the JAKAFI® group had some reduction in the spleen volume whereas majority of those in the placebo arm had increase in splenomegaly. There was a 46% reduction in the TSS (Total Symptom Score) at week 24 in the JAKAFI® group compared to 5% in the placebo group and majority of patients in the later group had worsening of symptoms (P<0.0001). When JAKAFI® was compared to Best Available Therapy (BAT) in the COMFORT-II study, 28% of the patients in the JAKAFI® group met the primary endpoint of a 35% or more reduction in the spleen volume at 48 weeks compared to none in the BAT group (P<0.0001). Over 55% had a mean decrease in spleen size in the JAKAFI® compared to a 4% mean increase in the BAT group. The 2 year follow up analyses from both these trials showed improved overall survival and a reduction in the risk of death for patients randomized to JAKAFI®, compared to those in the control groups. There was weight gain with alleviation of cachexia and improvements in splenomegaly and symptoms were durable. This benefit was seen in patients regardless of JAK mutations. It remains to be seen if JAKAFI® will benefit patients with Polycythemia Vera and Essential Thrombocythemia. Kantarjian HM, Silver RT, Komrokji RS, et al. Clinical Lymphoma Myeloma and Leukemia 2013; 13:638-645
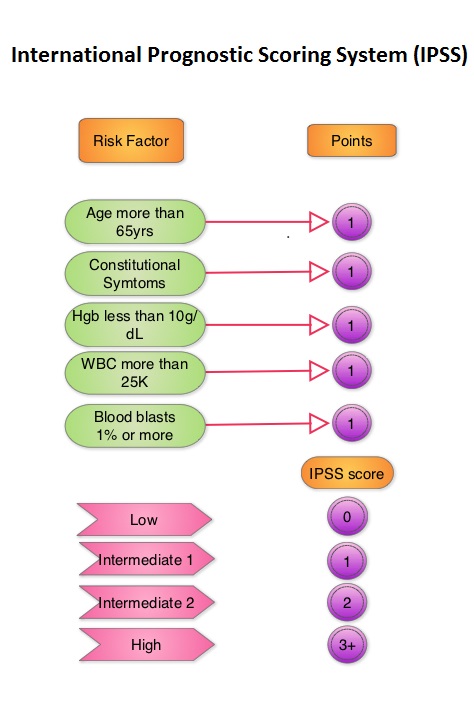
Randomized Open-Label Phase II Study of Decitabine in Patients With Low- or Intermediate-Risk Myelodysplastic Syndromes
SUMMARY: The MyeloDysplastic Syndromes (MDS) are disorders of hematopoietic stem cells, characterized by one or more peripheral blood cytopenias. These syndromes may arise de novo or can be secondary, following treatment with chemotherapy and/or radiation therapy for other malignancies. It can also manifest after environmental exposures. The International Prognostic Scoring System (IPSS), based on the score, divides patients with MDS into Lower Risk (IPSS low and intermediate-1 scores) and Higher Risk (IPSS intermediate-2 and high scores) groups. This classification remains arbitrary because of the heterogeneity in the Lower Risk group and poor outcomes in certain Lower Risk subset of patients. DACOGEN® (Decitabine) exerts its antineoplastic effects by inhibitng DNA methyltransferase, resulting in hypomethylation of DNA and apoptosis. The authors in this Phase II study evaluated the outcomes in patients with Low or Intermediate-1 risk MDS using lower doses of DACOGEN® given subcutaneously on two different treatment schedules. Of the 67 randomized patients, 43 patients received DACOGEN® 20mg/m2 SC QD for three consecutive days every 28 days (Schedule A) or 20mg/m2 SC given on days 1, 8 and 15, of a 28 day cycle (Schedule B). Treatment was planned for up to one year. Primary endpoint was Overall Improvement Rate (OIR) and this included Complete Remission (CR), Partial Remission (PR), marrow CR or Hematologic Improvement (HI). Secondary end points included HI, transfusion independence, cytogenetic response, overall survival (OS), and time to acute myeloid leukemia or death. Taken as a group, there was no difference in the OIR (23%), HI, transfusion independence or cytogenetic response between schedule A and schedule B. However, patients on schedule A had more CR's making this a protocol defined superior schedule. Median OS was not reached. Transfusion independency was noted in 67% of the patient's on schedule A and 59% of the patient's on schedule B. Further, approximately 70% of the patients were alive at 500 days. The most frequent adverse events in schedule A and B were anemia (23% vs 18%), neutropenia (28% vs 36%) and thrombocytopenia (16% vs 32%). Based on these promising results, the authors recommended DACOGEN® given on three consecutive days SC every 28 days as per Schedule A, for patients with Low or Intermediate-1 risk MDS. Garcia-Manero G, Jabbour E, Borthakur G, et al. J Clin Oncol 2013;31:2548-2553
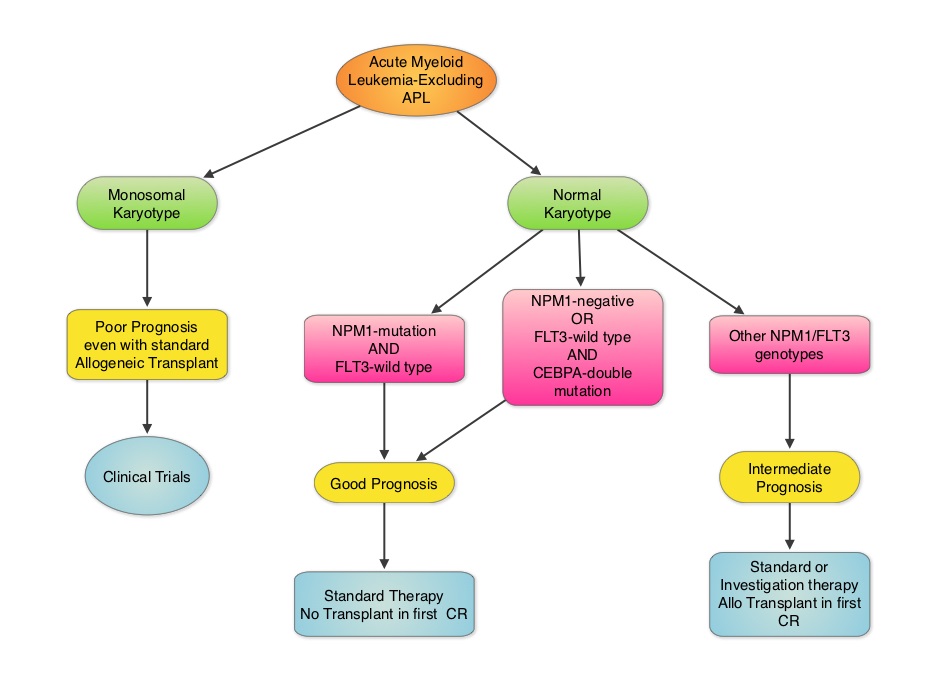
Effect of NPM1 and FLT3 Mutations on the Outcomes of Elderly Patients With Acute Myeloid Leukemia Receiving Standard Chemotherapy
SUMMARY: Cytogenetic analysis has been part of routine evaluation when caring for patients with AML. By predicting resistance to therapy, cytogenetics stratify patients based on risk and helps manage them accordingly. Even though normal karyotype is the most common cytogenetic finding, approximately 10%-15% of AML patients have a monosomal karyotype (presence of at least 2 autosomal monosomies or a single autosomal monosomy in combination with at least one structural abnormality). These patients have a poor prognosis and alterations in the TP53 gene has been implicated in majority of these patients. AML patients with a normal karyotype should be tested for NPM1 (Nucleophosmin), FLT3 (Fms-related tyrosine kinase 3) and CEBPA (CCAAT/Enhancer Binding Protein Alpha) mutations, in addition to cytogenetics, as this may have therapeutic implications. CEBPA is a transcription factor and plays an important role in myeloid differentiation. Mutations in the CEBPA gene have been described in approximately 10% of patients with AML. Patients can have one or two mutations in this gene. It appears that favorable outcomes may be limited to those patients who have double CEBPA mutations rather than those with single CEBPA mutations. AML patients without FLT3 mutations or NPM1 mutation with CEBPA-double mutations have a favorable outcome. In this retrospective review, the authors analyzed the clinical impact of NPM1 and FLT3 mutations in AML patients, 65 years of age or older, treated with cytotoxic chemotherapy. A total of 557 patients were retrospectively reviewed. They noted that the outcomes were significantly better amongst patients with NPM1-mut/FLT3-wild type genotype compared to any other NPM1/FLT3 genotypes. The median survival was 21.5 months vs. 9.0 months and estimated 2-year survival rates were 51% vs. 38%, respectively (P = .003). The authors concluded that elderly AML patients with NPM1-mut/FLT3-wild type genotype have significantly improved outcomes when treated with cytotoxic chemotherapy. This subset of patients have a good prognosis with outcomes similar to those with favorable cytogenetics such as Inversion 16 and t(8:21). The discovery of molecular mutations is providing valuable information to facilitate risk-adapted therapy. Daver N, Liu Dumlao T, Ravandi F, et al. Clinical Lymphoma Myeloma and Leukemia 2013;13:435-440
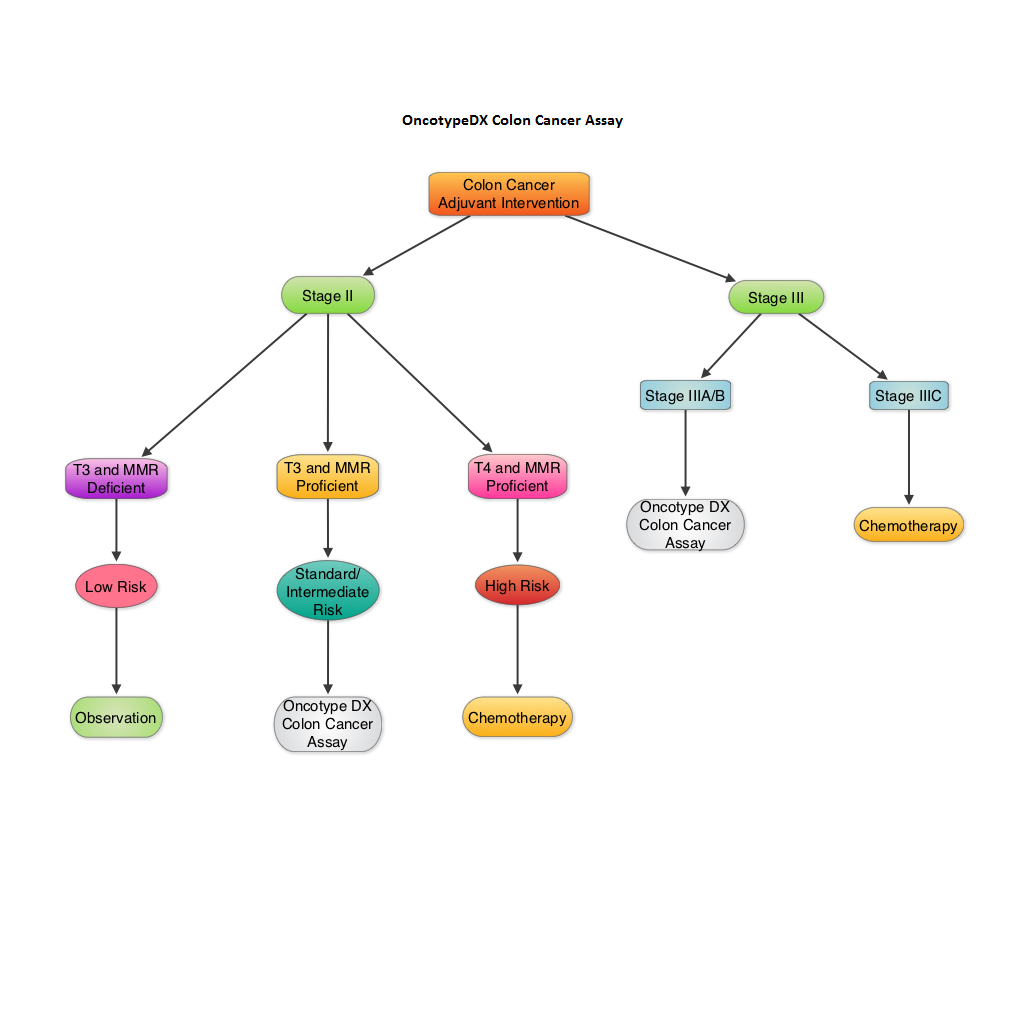
Validation of the 12-Gene Colon Cancer Recurrence Score in NSABP C-07 As a Predictor of Recurrence in Patients With Stage II and III Colon Cancer Treated With Fluorouracil and Leucovorin (FU/LV) and FU/LV Plus Oxaliplatin
SUMMARY: The beneficial role of adjuvant chemotherapy is established in Stage III colon Cancer. This however is less convincing in patients with stage II colon cancer. The conventional clinical and pathological features such as Tumor stage, Grade, Lymphovascular invasion and number of lymph nodes evaluated, may be of some prognostic significance in Stage II colon cancer, but have not been validated. Nonetheless, a majority of these patients receive ELOXATIN® (Oxaliplatin) based adjuvant chemotherapy, with very little benefit and significant toxicity. The 12-gene Colon Cancer Recurrence Score assay was developed after it was validated in multiple studies in over 3000 patients. This assay is not recommended for Stage II MMR (MisMatchRepair) Deficient patients, as these patients have a better survival and their tumors are resistant to 5-Fluorouracil and may actually do worse with adjuvant chemotherapy following surgery. The recurrent score corresponds to the likelihood of colon cancer recurrence 3 years after surgery, with a higher score suggesting a higher risk of recurrence. The authors in this analysis conducted a prospectively designed, clinical validation study of Recurrence Score, in 892 fixed Paraffin embedded tumor specimens from patients with stage II and III colon cancer, randomly assigned to Fluorouracil (FU) or FU plus ELOXATIN® in the National Surgical Adjuvant Breast and Bowel Project C-07. The primary objective of this study was to retrospectively determine the relationship between the recurrent score and the recurrent risk in stage II/III patients treated with 5-FU or 5-FU/ELOXATIN®. Secondary objectives were to determine if recurrence score provided significant information beyond T stage, tumor grade, MMR status and number of lymph nodes examined. From this analysis, the authors reported that recurrent score performs similarly in both Stage II and Stage III colon cancer patients and predicts recurrence risk beyond T and N stage, Grade, MMR status, number of lymph nodes examined and treatment. Amongst Stage II patients, T4 tumors that are MMR proficient fall in the high risk category and chemotherapy should be considered regardless of the recurrent score whereas T3 tumors that are MMR proficient fall in the standard/intermediate risk category and recurrent score assay may assist in the decision making process. In this group, a recurrence score of 41 or more puts these patients at a recurrence risk that is similar to those who have T4 tumors that is MMR proficient and will therefore benefit from adjuvant chemotherapy. In certain stage IIIA/B patients, a low recurrence score of less than 30 is associated with low absolute benefit with ELOXATIN® and therefore is not recommended. The authors concluded that the 12- Gene Colon Cancer Recurrent Score results can help guide adjuvant therapy decisions for patients with Stage II colon cancer and certain patients with Stage III colon cancer. Yothers G, O’Connell MJ, Lee M, et al. J Clin Oncol 2013;31:4512-4519.
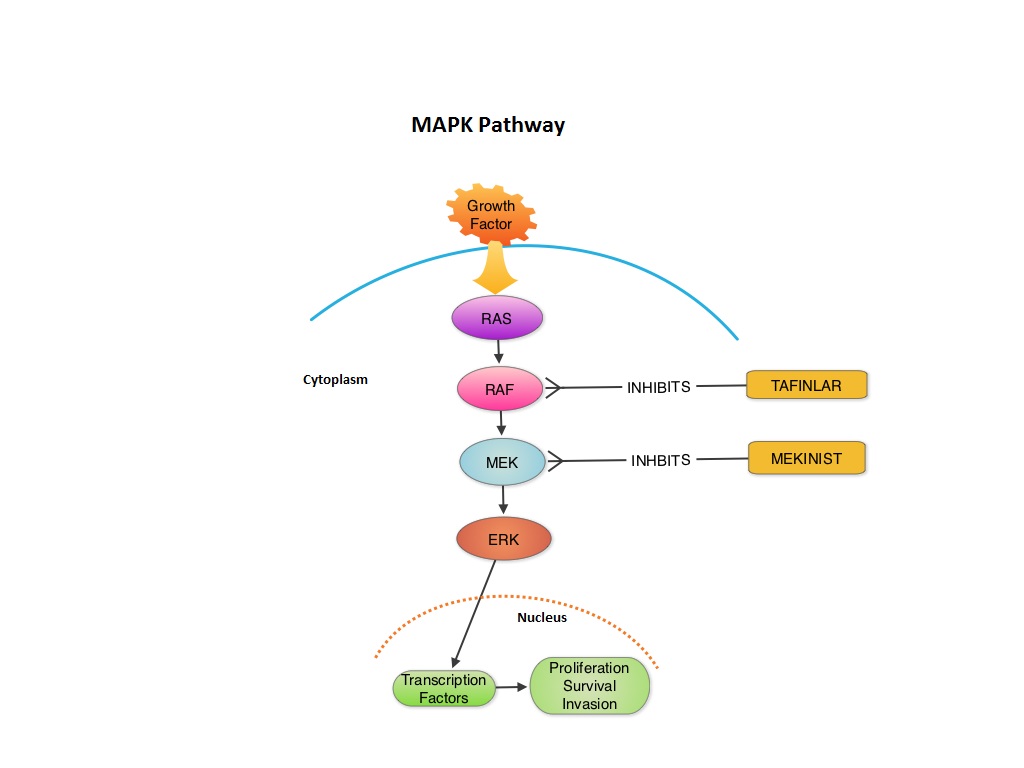
Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations
SUMMARY:The FDA granted accelerated approval in January 2014, for a combination of MEKINIST® (Trametinib) and TAFINLAR® (Dabrafenib), to treat patients with advanced melanoma, based on the understanding of the biological pathways of this malignancy. The Mitogen-Activated Protein Kinase pathway (MAPK pathway) is an important signaling pathway which enables the cell to respond to external stimuli. This pathway plays a dual role regulating cytokine production and participating in cytokine dependent signaling cascade. The MAPK pathway of interest is the RAS-RAF-MEK-ERK pathway. The RAF family of kinases includes ARAF, BRAF and CRAF signaling molecules. BRAF is a very important intermediary of the RAS-RAF-MEK-ERK pathway. BRAF mutations have been demonstrated in 6%-8% of all malignancies. The most common BRAF mutation in melanoma is at the V600E site and is detected in approximately 50% of melanomas. In the BREAK-3 randomized phase III trial, TAFINLAR®, a selective oral BRAF inhibitor demonstrated a statistically significant improvement in Progression Free Survival (PFS) and Response Rate (RR) compared to Dacarbazine (DTIC) in patients with advanced BRAF V600E mutated melanoma. Squamous cell carcinoma’s were seen in 6% of the patients. In the METRIC phase III study, MEKINIST®, a potent and selective inhibitor of MEK gene (which is downstream from RAF in the MAPK pathway) was compared with either Dacarbazine or TAXOL® (Paclitaxel) in advanced melanoma patients with BRAF V600E/K mutations. Patients in the MEKINIST® group had a significantly improved PFS, RR and Overall Survival. Based on the understanding of the biological pathways of the disease and different mechanisms of action (MOA) of these two agents, a phase I and II trial was conducted combining TAFINLAR® and MEKINIST®. In this study, 162 treatment naïve patients with unresectable or metastatic melanoma, with BRAF V600E or V600K mutations received either TAFINLAR® 150 mg plus MEKINIST® 1 or 2 mg or TAFINLAR® alone. Treatment was given until disease progression or side effects were intolerable. The primary end points were the incidence of Cutaneous Squamous-cell carcinoma, PFS and RR. Secondary end points included Overall Survival and pharmacokinetic activity. The combination treatment resulted in a lower incidence of Cutaneous Squamous-cell carcinoma (7% vs 19% in those receiving monotherapy, P=0.09), improved median PFS (9.4 months vs 5.8 months in the monotherapy group, HR=0.39, P<0.001) and superior complete or partial responses (76% compared with 54% with monotherapy (P=0.03). Pyrexia was however more common in the combination group than in the monotherapy group (71% vs. 26%). The authors concluded that by combining two agents with different MOA’s targeting two different tyrosine kinases in the RAS/RAF/MEK/ERK pathway, PFS was significantly improved. Further, development of resistance can be overcome in BRAF mutation positive melanoma patients and the incidence of secondary skin cancers found with BRAF inhibitor monotherapy can be reduced as well. Flaherty KT, Infante JR, Daud A, et al. N Engl J Med 2012;367:1694-1703
Multiple Myeloma International Staging System “Staging” or Simply “Aging” System?
SUMMARY: In this provocative perspective, the authors suggest revising the present Multiple Myeloma (MM) International Staging System, to incorporate more relevant information related to the biology of the disease. The MM International Staging System (MM-ISS) takes into account Beta-2 Microglobulin (B2M) along with Serum Albumin (SA) levels to determine the prognosis in MM patients. 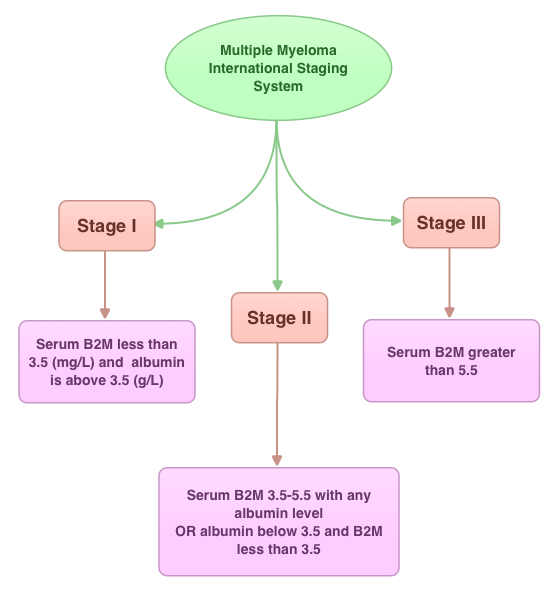 Although B2M and SA may act as surrogates for the extent of the disease, the evidence is insufficient to rely on these two entities to prognosticate MM. Even though B2M can correlate with MM tumor bulk as well as act as a biomarker of renal failure related to MM, the authors cite multiple studies suggesting that elevated serum B2M can be seen independent of MM history, in healthy elderly patients, infections, autoimmune disorders and chronic renal insufficiency. Further elevated B2M can be a marker of frailty in elderly patients and can predict disability and mortality in this patient group. With regards to Serum Albumin (SA) in MM, SA is inhibited by IL6, a MM cell growth factor and thus is able to indirectly reflect MM tumor bulk. However, low SA levels can also be seen in frail and elderly patients independent of MM history. The authors point out that the stage of the disease in the MM-ISS increases with age, independent of MM tumor bulk, due to the these nonspecific factors and does not necessarily correlate with other more specific prognostic markers such as cytogenetics, particularly in elderly patients. The authors conclude that combining the MM-ISS with tumor cytogenetics will more accurately predict prognosis in MM patients. Bataille R, Annweiler C, Beauchet O. Clinical Lymphoma Myeloma and Leukemia 2013;13:635-637
Although B2M and SA may act as surrogates for the extent of the disease, the evidence is insufficient to rely on these two entities to prognosticate MM. Even though B2M can correlate with MM tumor bulk as well as act as a biomarker of renal failure related to MM, the authors cite multiple studies suggesting that elevated serum B2M can be seen independent of MM history, in healthy elderly patients, infections, autoimmune disorders and chronic renal insufficiency. Further elevated B2M can be a marker of frailty in elderly patients and can predict disability and mortality in this patient group. With regards to Serum Albumin (SA) in MM, SA is inhibited by IL6, a MM cell growth factor and thus is able to indirectly reflect MM tumor bulk. However, low SA levels can also be seen in frail and elderly patients independent of MM history. The authors point out that the stage of the disease in the MM-ISS increases with age, independent of MM tumor bulk, due to the these nonspecific factors and does not necessarily correlate with other more specific prognostic markers such as cytogenetics, particularly in elderly patients. The authors conclude that combining the MM-ISS with tumor cytogenetics will more accurately predict prognosis in MM patients. Bataille R, Annweiler C, Beauchet O. Clinical Lymphoma Myeloma and Leukemia 2013;13:635-637