Expert opinion: Luis Raez, MD, FACP, FCCP, FASCO
Content sponsored by Bristol Myers Squibb
OPDIVO® (nivolumab) + YERVOY® (ipilimumab) + 2 cycles of platinum-doublet chemotherapy in 1L mNSCLC
Checkmate 9LA, a randomized, open-label, phase 3 trial, led to the approval of OPDIVO + YERVOY + chemotherapy as a treatment for 1L r/m NSCLC with no EGFR or ALK genomic tumor aberrations and regardless of PD-L1 status.1,3‡ “In my practice, I see a large number of patients with PD-L1 <1% mNSCLC and, in general, PD-L1 <1% has a worse prognosis than PD-L1 >1%,” stated Dr Raez.
*In Checkmate 9LA, patients received 2 cycles of platinum-doublet chemo q3w in the experimental arm and 4 cycles in the comparator arm; NSQ: pemetrexed + carboplatin or cisplatin (optional pemetrexed maintenance therapy in the comparator arm only); SQ: paclitaxel + carboplatin.1 †Dr Raez was compensated by BMS for his contributions to this article. ‡Although PD-L1 status was not a restriction on the trial’s eligibility criteria, it is not part of the approved indication.1
OPDIVO and YERVOY are associated with the following Warnings and Precautions: severe and fatal immune-mediated adverse reactions including pneumonitis, colitis, hepatitis and hepatotoxicity, endocrinopathies, nephritis with renal dysfunction, dermatologic adverse reactions, other immune-mediated adverse reactions; infusion-related reactions; complications of allogeneic hematopoietic stem cell transplantation (HSCT); embryo-fetal toxicity; and increased mortality in patients with multiple myeloma when OPDIVO is added to a thalidomide analogue and dexamethasone, which is not recommended outside of controlled clinical trials.
Please see additional Important Safety Information for OPDIVO and YERVOY below, and U.S. Full Prescribing Information for OPDIVO and YERVOY.
The trial design of Checkmate 9LA included enrolling 719 eligible patients randomized 1:1 to receive either OPDIVO 360 mg q3w + YERVOY 1 mg/kg q6w + 2 cycles of platinum-doublet chemotherapy q3w (n=361) or platinum-doublet chemotherapy alone q3w (n=358).1 Key eligibility criteria included age of 18 years or older, stage IV or recurrent NSCLC, ECOG PS 0/1, and no prior systemic anticancer therapy.1 Treatment continued until disease progression, unacceptable toxicity, or for up to 2 years.1 Patients were stratified by histology (SQ vs NSQ), PD-L1 status (<1% vs ≥1%), and sex.4 The primary endpoint was OS and additional efficacy outcome measures were PFS, ORR, and DOR.3
Checkmate 9LA was the first phase 3 study to demonstrate improved OS regardless of PD-L1 expression.4 Furthermore, this is the only I-O combination with more than 1 in 5 patients with PD-L1 <1% alive at 5 years.1,3 A limitation to note is that Checkmate 9LA was not powered to detect differences in treatment effect in PD-L1 subgroups; therefore, results from this exploratory analysis should be interpreted with caution due to the limited patient numbers and potential imbalances in baseline characteristics within the subgroup.
In the primary analysis (minimum follow-up of 8.1 months), OPDIVO + YERVOY and chemotherapy demonstrated4:
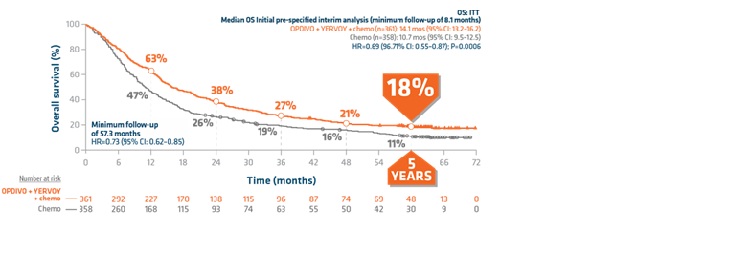
- Statistically significant and superior mOS (14.1 months; [95% CI: 13.2–16.2]) vs chemotherapy alone (10.7 months; [95% CI: 9.5–12.5]) in the ITT population (HR=0.69; [96.71% CI: 0.55–0.87]; P=0.0006)1
- Improved overall survival5‡:
- PD-L1 <1% patient population (14.0 months with OPDIVO + YERVOY and chemo [95% CI: 13.2–NR] vs 10.0 months with chemotherapy alone [95% CI: 7.7–13.7])
- PD-L1 ≥1% patient population (14.2 months with OPDIVO + YERVOY and chemo [95% CI: 13.1–NR] vs 10.6 with chemotherapy alone [95% CI: 9.4–12.6])
‡Limitation: Checkmate 9LA was not powered to detect differences in treatment effect in PD-L1 subgroups; therefore, results from this exploratory analysis should be interpreted with caution due to the limited patient numbers and potential imbalances in baseline characteristics within the subgroup.
In a 5-year follow-up analysis, durable survival and continued response to treatment were observed with OPDIVO + YERVOY and 2 cycles of chemotherapy compared with chemotherapy alone. The following data were observed at the 57.3-month minimum follow-up3:
- mOS3:
- ITT patient population: 15.8 months (95% CI: 13.9–19.7) with OPDIVO + YERVOY and chemo vs 11.0 months (95% CI: 9.5–12.7) with chemo (HR=0.73; [95% CI: 0.62–0.85])
- PD-L1 <1% patient population: 17.7 months (95% CI: 13.7–20.3) with OPDIVO + YERVOY and chemo vs 9.8 months (95% CI: 7.7–13.5) with chemo (HR=0.63; [95% CI: 0.49–0.83])
- PD-L1 ≥1% patient population: 15.8 months (95% CI: 13.8–22.2) with OPDIVO + YERVOY and chemo vs 10.9 months (95% CI: 9.5–13.2) with chemo (HR=0.73; [95% CI: 0.59–0.90])
Durable overall survival rate in patients with PD-L1 <1%: The only I-O combination with more than 1 in 5 patients alive at 5 years1,3,5,6
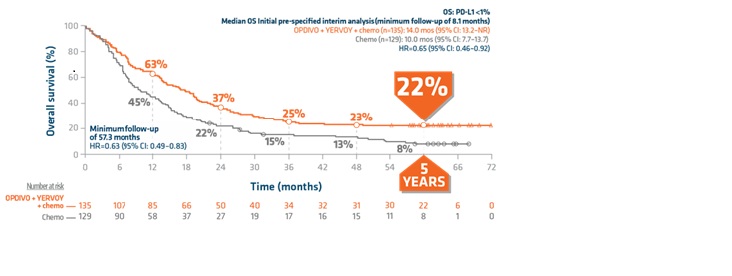
Limitation: Checkmate 9LA was not powered to detect differences in treatment effect in PD-L1 subgroups; therefore, results from this exploratory analysis should be interpreted with caution due to the limited patient numbers and potential imbalances in baseline characteristics within the subgroup.
“22% is high compared with 8% and that is why we like the Checkmate 9LA regimen,” explained Dr Raez.
Overall survival in ITT population: Extended 5-year follow-up analysis1,3,6
- mDOR3:
- PD-L1 <1% patient population: 17.5 months with OPDIVO + YERVOY and chemo (95% CI: 6.9–37.8) vs 4.3 months with chemo (95% CI: 2.8–7.1)
- PD-L1 ≥1% patient population: 11.8 months (95% CI: 8.6–20.3) vs 5.6 months and chemo (95% CI: 4.3–8.0)
- ITT patient population: 12.4 months with OPDIVO + YERVOY and chemo (95% CI: 8.7–20.2) vs 5.6 months with chemo (95% CI: 4.4–7.1)
Duration of response in PD-L1 <1%: Extended 5-year follow-up analysis3
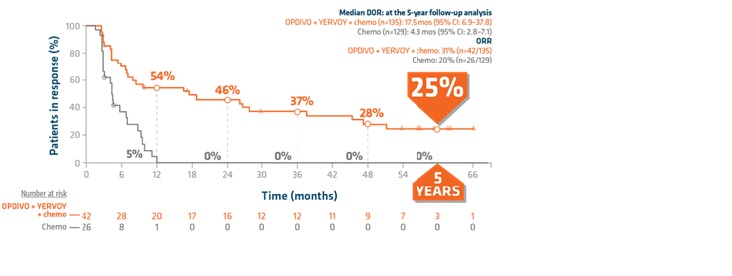
Limitation: Checkmate 9LA was not powered to detect differences in the treatment effect in this subgroup;
therefore, this exploratory analysis should be interpreted with caution because of the limited patient
numbers and potential imbalances in baseline characteristics within the subgroup.
“This regimen has markedly prolonged the duration of response at 5 years for 25% of patients with PD-L1 <1%” stated Dr. Raez.
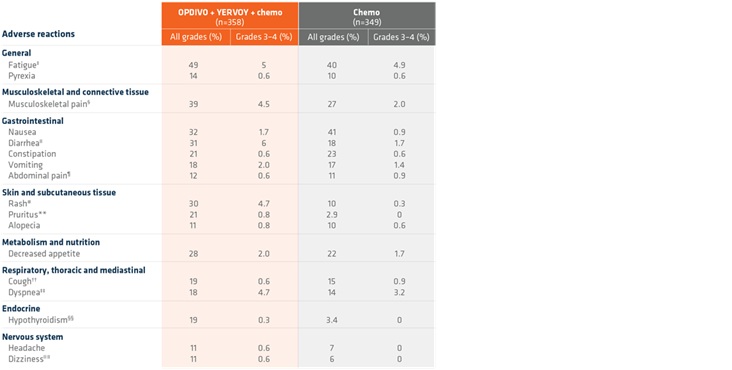
Adverse reactions in >10% of patients receiving OPDIVO + YERVOY and 2 cycles of chemo1*†
- OPDIVO + YERVOY with chemo was discontinued in 24% of patients due to adverse reactions, and 56% had at least one dose withheld for an adverse reaction1
- Serious adverse reactions occurred in 57% of patients receiving OPDIVO + YERVOY with chemo1
- The most frequent (>2%) serious adverse reactions were pneumonia, diarrhea, febrile neutropenia, anemia, acute kidney injury, musculoskeletal pain, dyspnea, pneumonitis, and respiratory failure. Fatal adverse reactions occurred in 7 (2%) patients, and included hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia1
- The most common (>20%) adverse reactions were fatigue, musculoskeletal pain, nausea, diarrhea, rash, decreased appetite, constipation, and pruritus1
- Median number of doses was 9 for OPDIVO, 4 for YERVOY, and 2 cycles of chemo7
- With a minimum follow-up of 57.3 months, no new safety signals were identified for OPDIVO + YERVOY and 2 cycles of chemo3*
Toxicity was graded per NCI CTCAE v4.1
*vs chemo. In Checkmate 9LA, patients received 2 cycles of platinum-doublet chemo q3w in the experimental arm and 4 cycles in the comparator arm; NSQ: pemetrexed + carboplatin or cisplatin (optional pemetrexed maintenance therapy in the comparator arm only); SQ: paclitaxel + carboplatin.1 †Based on types of adverse reactions reported in 1L mNSCLC. Please note clinical trials are conducted under varying conditions, including different trial designs and dosing. Adverse reaction rates cannot be directly compared between trials.1 ‡Includes fatigue and asthenia.1 §Includes myalgia, back pain, pain in extremity, musculoskeletal pain, bone pain, flank pain, muscle spasms, musculoskeletal chest pain, musculoskeletal disorder, osteitis, musculoskeletal stiffness, non-cardiac chest pain, arthralgia, arthritis, arthropathy, joint effusion, psoriatic arthropathy, and synovitis.1 ║Includes colitis, ulcerative colitis, diarrhea, and enterocolitis.1 ¶Includes abdominal discomfort, abdominal pain, lower abdominal pain, upper abdominal pain, and gastrointestinal pain.1 #Includes acne, dermatitis, acneiform dermatitis, allergic dermatitis, atopic dermatitis, bullous dermatitis, generalized exfoliative dermatitis, eczema, keratoderma blennorrhagica, palmar-plantar erythrodysesthesia syndrome, rash, erythematous rash, generalized rash, macular rash, maculo-papular rash, morbilliform rash, papular rash, pruritic rash, skin exfoliation, skin reaction, skin toxicity, Stevens-Johnson syndrome, and urticaria.1 **Includes pruritus and generalized pruritus.1 ††Includes cough, productive cough, and upper-airway cough syndrome.1 ‡‡Includes dyspnea, dyspnea at rest, and exertional dyspnea.1 §§Includes autoimmune thyroiditis, increased blood thyroid stimulating hormone, hypothyroidism, thyroiditis, and decreased free tri-iodothyronine.1 ║║Includes dizziness, vertigo and positional vertigo.1
“The safety profile in the extended 5-year follow-up analysis of Checkmate 9LA was consistent with the previously known profiles for each component,” explained Dr Raez.
Dosing
OPDIVO® (nivolumab) + low-dose YERVOY® (ipilimumab) (1 mg/kg) and 2 cycles of chemo1
For the r/m NSCLC dosing regimen in combination with chemo: on the first week, 4 agents will be administered (OPDIVO 360 mg + YERVOY 1 mg/kg + platinum-doublet histology-based chemo†), followed by 3 agents (OPDIVO + platinum-doublet histology-based chemo†) on the third week, 2 agents (OPDIVO + YERVOY) on the sixth week, and OPDIVO monotherapy on the ninth week, followed by maintenance therapy of OPDIVO + YERVOY: OPDIVO 360 mg q3w + YERVOY 1 mg/kg q6w until disease progression, unacceptable toxicity, or for up to 2 years.1 Histology-based chemo: SQ patients: carboplatin AUC 6 + paclitaxel 200 mg/m2 q3w; NSQ patients: carboplatin AUC 5 or 6 or cisplatin 75 mg/m2 + pemetrexed 500 mg/m2 q3w. No chemo maintenance required.1
- OPDIVO is administered as an IV infusion over 30 minutes1
- YERVOY is administered as an IV infusion over 30 minutes2
Summary and conclusions
5-year follow-up analysis of Checkmate 9LA continues to show prolonged survival data with OPDIVO + YERVOY and chemo vs chemo alone in patients who have r/m NSCLC across PD-L1 <1% and ≥1% expression.1,3 “Checkmate 9LA shows patient survival data that may be impactful,” stated Dr. Raez.
1L=first line; ALK=anaplastic lymphoma kinase; AUC=area under the curve; CI=confidence interval; DOR=duration of response; ECOG PS=Eastern Cooperative Oncology Group Performance Status; EGFR=epidermal growth factor receptor; HR=hazard ratio; I-O=immuno-oncology; ITT=intent to treat; IV=intravenous; mDOR=median DOR; mNSCLC=metastatic NSCLC; mo=month; mOS=median OS; NR=not reached; NSCLC=non-small cell lung cancer; NSQ=non-squamous; ORR=overall response rate; OS=overall survival; PD-1=programmed death receptor-1; PD-L1=programmed death ligand 1; PFS=progression-free survival; Pt=platinum; q3w=every 3 weeks; q6w=every 6 weeks; r/m=recurrent or metastatic; SQ=squamous.
INDICATION
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
IMPORTANT SAFETY INFORMATION
Severe and Fatal Immune-Mediated Adverse Reactions
- Immune-mediated adverse reactions listed herein may not include all possible severe and fatal immune- mediated adverse reactions.
- Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue. While immune-mediated adverse reactions usually manifest during treatment, they can also occur after discontinuation of OPDIVO or YERVOY. Early identification and management are essential to ensure safe use of OPDIVO and YERVOY. Monitor for signs and symptoms that may be clinical manifestations of underlying immune-mediated adverse reactions. Evaluate clinical chemistries including liver enzymes, creatinine, adrenocorticotropic hormone (ACTH) level, and thyroid function at baseline and periodically during treatment with OPDIVO and before each dose of YERVOY. In cases of suspected immune-mediated adverse reactions, initiate appropriate workup to exclude alternative etiologies, including infection. Institute medical management promptly, including specialty consultation as appropriate.
- Withhold or permanently discontinue OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information). In general, if OPDIVO or YERVOY interruption or discontinuation is required, administer systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) until improvement to Grade 1 or less. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Consider administration of other systemic immunosuppressants in patients whose immune-mediated adverse reactions are not controlled with corticosteroid therapy. Toxicity management guidelines for adverse reactions that do not necessarily require systemic steroids (e.g., endocrinopathies and dermatologic reactions) are discussed below.
Immune-Mediated Pneumonitis
- OPDIVO and YERVOY can cause immune-mediated pneumonitis. The incidence of pneumonitis is higher in patients who have received prior thoracic radiation.
Immune-Mediated Colitis
- OPDIVO and YERVOY can cause immune-mediated colitis, which may be fatal. A common symptom included in the definition of colitis was diarrhea. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-mediated colitis. In cases of corticosteroid-refractory colitis, consider repeating infectious workup to exclude alternative etiologies.
Immune-Mediated Hepatitis and Hepatotoxicity
- OPDIVO and YERVOY can cause immune-mediated hepatitis.
Immune-Mediated Endocrinopathies
- OPDIVO and YERVOY can cause primary or secondary adrenal insufficiency, immune-mediated hypophysitis, immune-mediated thyroid disorders, and Type 1 diabetes mellitus, which can present with diabetic ketoacidosis. Withhold OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information). For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Hypophysitis can present with acute symptoms associated with mass effect such as headache, photophobia, or visual field defects. Hypophysitis can cause hypopituitarism; initiate hormone replacement as clinically indicated. Thyroiditis can present with or without endocrinopathy. Hypothyroidism can follow hyperthyroidism; initiate hormone replacement or medical management as clinically indicated. Monitor patients for hyperglycemia or other signs and symptoms of diabetes; initiate treatment with insulin as clinically indicated.
Immune-Mediated Nephritis with Renal Dysfunction
- OPDIVO and YERVOY can cause immune-mediated nephritis.
Immune-Mediated Dermatologic Adverse Reactions
- OPDIVO can cause immune-mediated rash or dermatitis. Exfoliative dermatitis, including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug rash with eosinophilia and systemic symptoms (DRESS) has occurred with PD-1/PD-L1 blocking antibodies. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate nonexfoliative rashes.
- YERVOY can cause immune-mediated rash or dermatitis, including bullous and exfoliative dermatitis, SJS, TEN, and DRESS. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate non-bullous/exfoliative rashes.
- Withhold or permanently discontinue OPDIVO and YERVOY depending on severity (please see section 2 Dosage and Administration in the accompanying Full Prescribing Information).
Other Immune-Mediated Adverse Reactions
- The following clinically significant immune-mediated adverse reactions occurred at an incidence of <1% (unless otherwise noted) in patients who received OPDIVO monotherapy or OPDIVO in combination with YERVOY or were reported with the use of other PD-1/PD-L1 blocking antibodies. Severe or fatal cases have been reported for some of these adverse reactions: cardiac/vascular: myocarditis, pericarditis, vasculitis; nervous system: meningitis, encephalitis, myelitis and demyelination, myasthenic syndrome/myasthenia gravis (including exacerbation), Guillain-Barré syndrome, nerve paresis, autoimmune neuropathy; ocular: uveitis, iritis, and other ocular inflammatory toxicities can occur; gastrointestinal: pancreatitis to include increases in serum amylase and lipase levels, gastritis, duodenitis; musculoskeletal and connective tissue: myositis/polymyositis, rhabdomyolysis, and associated sequelae including renal failure, arthritis, polymyalgia rheumatica; endocrine: hypoparathyroidism; other (hematologic/immune): hemolytic anemia, aplastic anemia, hemophagocytic lymphohistiocytosis (HLH), systemic inflammatory response syndrome, histiocytic necrotizing lymphadenitis (Kikuchi lymphadenitis), sarcoidosis, immune thrombocytopenic purpura, solid organ transplant rejection, other transplant (including corneal graft) rejection.
- In addition to the immune-mediated adverse reactions listed above, across clinical trials of YERVOY monotherapy or in combination with OPDIVO, the following clinically significant immune-mediated adverse reactions, some with fatal outcome, occurred in <1% of patients unless otherwise specified: nervous system: autoimmune neuropathy (2%), myasthenic syndrome/myasthenia gravis, motor dysfunction; cardiovascular: angiopathy, temporal arteritis; ocular: blepharitis, episcleritis, orbital myositis, scleritis; gastrointestinal: pancreatitis (1.3%); other (hematologic/immune): conjunctivitis, cytopenias (2.5%), eosinophilia (2.1%), erythema multiforme, hypersensitivity vasculitis, neurosensory hypoacusis, psoriasis.
- Some ocular IMAR cases can be associated with retinal detachment. Various grades of visual impairment, including blindness, can occur. If uveitis occurs in combination with other immune-mediated adverse reactions, consider a Vogt-Koyanagi-Harada–like syndrome, which has been observed in patients receiving OPDIVO and YERVOY, as this may require treatment with systemic corticosteroids to reduce the risk of permanent vision loss.
Infusion-Related Reactions
- OPDIVO and YERVOY can cause severe infusion-related reactions. Discontinue OPDIVO and YERVOY in patients with severe (Grade 3) or life-threatening (Grade 4) infusion-related reactions. Interrupt or slow the rate of infusion in patients with mild (Grade 1) or moderate (Grade 2) infusion-related reactions.
Complications of Allogeneic Hematopoietic Stem Cell Transplantation
- Fatal and other serious complications can occur in patients who receive allogeneic hematopoietic stem cell transplantation (HSCT) before or after being treated with OPDIVO or YERVOY. Transplant-related complications include hyperacute graft-versus-host-disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease (VOD) after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause). These complications may occur despite intervening therapy between OPDIVO or YERVOY and allogeneic HSCT.
- Follow patients closely for evidence of transplant-related complications and intervene promptly. Consider the benefit versus risks of treatment with OPDIVO and YERVOY prior to or after an allogeneic HSCT.
Embryo-Fetal Toxicity
- Based on its mechanism of action and findings from animal studies, OPDIVO and YERVOY can cause fetal harm when administered to a pregnant woman. The effects of YERVOY are likely to be greater during the second and third trimesters of pregnancy. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with OPDIVO and YERVOY and for at least 5 months after the last dose.
Increased Mortality in Patients with Multiple Myeloma when OPDIVO is Added to a Thalidomide Analogue and Dexamethasone
- In randomized clinical trials in patients with multiple myeloma, the addition of OPDIVO to a thalidomide analogue plus dexamethasone resulted in increased mortality. Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials.
Lactation
- There are no data on the presence of OPDIVO or YERVOY in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment and for 5 months after the last dose.
Serious Adverse Reactions
- In Checkmate 9LA, serious adverse reactions occurred in 57% of patients (n=358). The most frequent (>2%) serious adverse reactions were pneumonia, diarrhea, febrile neutropenia, anemia, acute kidney injury, musculoskeletal pain, dyspnea, pneumonitis, and respiratory failure. Fatal adverse reactions occurred in 7 (2%) patients, and included hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.
Common Adverse Reactions
- In Checkmate 9LA, the most common (>20%) adverse reactions were fatigue (49%), musculoskeletal pain (39%), nausea (32%), diarrhea (31%), rash (30%), decreased appetite (28%), constipation (21%), and pruritus (21%).
Please see US Full Prescribing Information for OPDIVO and YERVOY.
References:
- OPDIVO [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
- YERVOY [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
- Reck M, Ciuleanu TE, Schenker M, et al. Five-year outcomes with first-line nivolumab plus ipilimumab with 2 cycles of chemotherapy versus 4 cycles of chemotherapy alone in patients with metastatic non-small cell lung cancer in the randomized CheckMate 9LA trial. Eur J Cancer. Published online August 25, 2024. doi:10.1016/j.ejca.2024.114296
- Paz-Ares L, Ciuleanu TE, Cobo M, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(2):198-211.
- Data on file. NIVO 566. Princeton, NJ: Bristol-Myers Squibb Company; 2020.
- Reck M, Ciuleanu T-E, Cobo M, et al. First-line nivolumab plus ipilimumab with two cycles of chemotherapy versus chemotherapy alone (four cycles) in advanced non-small cell lung cancer: CheckMate 9LA 2-year update. ESMO Open. 2021;6(5):100273.
- Data on file. NIVO 562. Princeton NJ: Bristol-Myers Squibb Company; 2020.
© 2024 Bristol-Myers Squibb Company. OPDIVO® and YERVOY® are registered trademarks of Bristol-Myers Squibb Company.
7356-US-2400513 12/24
