Expert opinion: Jyoti Malhotra, MD, MPH
Content sponsored by: Bristol Myers Squibb
Dr. Malhotra was compensated by BMS for her contributions to this article.
Introduction: Unmet need in ROS1+ NSCLC
The identification of ROS1 as a therapeutic target in NSCLC has led to the development and approval of several first-generation TKIs.3-5 Despite this, the median duration of response is ~2 years with these first-generation TKIs.6,7 A different approach is needed.
1L AUGTYRO in locally advanced or metastatic ROS1+ NSCLC
TRIDENT-1, a global, phase 1/2, single-arm, multicohort, open-label trial, led to the approval of AUGTYRO as a treatment option in adult patients for locally advanced or metastatic ROS1+ NSCLC.1,2,8 AUGTYRO is the first and only approved next-generation TKI for this indication.8,9 “This approval has made it possible for newly diagnosed patients to have access to [another] treatment option that may provide disease control,” stated Dr. Malhotra.
TRIDENT-1 Trial Design1,3,10,11 In TRIDENT-1, the phase 2 dose expansion cohort included 127 patients who were either TKI-naïve (n=71) or had received a TKI (n=56).2 The primary endpoint was ORR and some of the secondary efficacy outcome measures were DOR and intracranial response. Baseline characteristics were reported for patients who had and had not received a prior TKI.2
In TRIDENT-1, the phase 2 dose expansion cohort included 127 patients who were either TKI-naïve (n=71) or had received a TKI (n=56).2 The primary endpoint was ORR and some of the secondary efficacy outcome measures were DOR and intracranial response. Baseline characteristics were reported for patients who had and had not received a prior TKI.2
There are warnings and precautions associated with AUGTYRO to keep in mind. These include central nervous system adverse reactions, interstitial lung disease (ILD)/pneumonitis, hepatotoxicity, myalgia with creatinine phosphokinase (CPK) elevation, hyperuricemia, skeletal fractures, and embryo-fetal toxicity.1 Additional information related to warnings and precautions can be found here.
In the primary analysis, efficacy results for the TKI-naïve population (n=71) treated with AUGTYRO were as follows12:
- ORR of 79% ([95% CI: 68–88]; median follow-up for ORR data: 18.1 months)
- CR of 6% (n=4)
- PR of 73% (n=52)
- mDOR of 34 months ([95% CI: 25.6–NE]; range: 1.4+ to 42.4+ months; median follow-up for DOR data: 24.0 months)1,10
- icORR observed in 7/8 patients with measurable baseline brain metastasis (median follow-up for icORR data: 18.1 months)1,12
Change in tumor burden by BICR in the TKI-naïve population12*
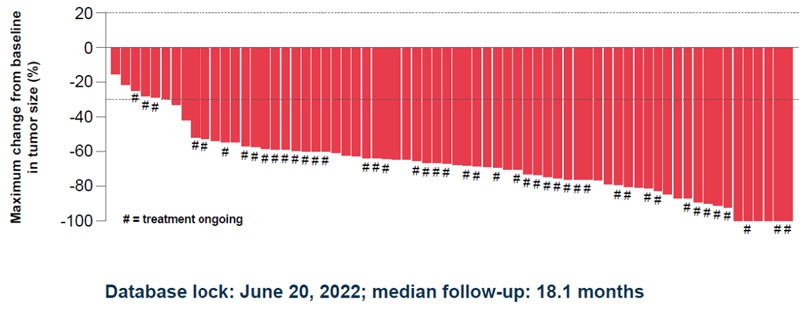
*Three patients discontinued study treatment before completing any post-baseline scans.12
In a follow-up analysis, continued response to treatment was seen with AUGTYRO. At the 33.9-month median follow-up, efficacy results for the TKI-naïve population treated with AUGTYRO were as follows9:
• cORR of 79% (n=71; [95% CI: 68–88])
• mDOR of 34.1 months (n=71; [95% CI: 27.4–NE])
• icORR of 89% (n=9; [95% Cl: 52–100]) in patients with measurable baseline brain metastasis
“TRIDENT-1 demonstrated an ORR of 79%, but more notably, a long mDOR of 34 months—this is almost 3 years,” explained Dr. Malhotra.
In TRIDENT-1, the most common reactions reported in ≥20% of 426 patients treated with AUGTYRO at the recommended dose were dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and cognitive impairment.1 AUGTYRO was discontinued in 7% of patients, interrupted in 50% of patients, and dosage was reduced in 38% of patients due to adverse reactions.1 Serious adverse reactions occurred in 35% of patients receiving AUGTYRO. The most frequent (≥2%) serious adverse reactions were pneumonia, dyspnea, pleural effusion, and hypoxia. Fatal adverse reactions occurred in 3.5% of patients and included pneumonia, pneumonia aspiration, cardiac arrest, sudden cardiac death, cardiac failure, hypoxia, dyspnea, respiratory failure, tremor, and disseminated intravascular coagulation.1
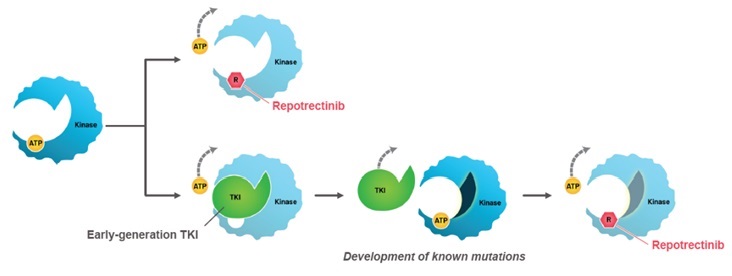
AUGTYRO is a next-generation ROS1 TKI with a compact structure that is smaller than currently available ROS1 TKIs.1,13,14
• Potential to decrease the development of ROS1 resistance mutations
• Potential to circumvent known ROS1 resistance mutations
• Physiochemical parameters for enhanced intracranial activity
Mechanism of Action1,13
Dosing of AUGTYRO
The recommended oral dose of AUGTYRO is1:
• 160 mg (4x 40-mg capsules, or a single 160-mg capsule, QD) for the first 14 days
• 160 mg (4x 40-mg capsules, or a single 160-mg capsule, BID) on Day 15 and onward, until disease progression or unacceptable toxicity
“More recently, the 160 mg tablet is also available for use, which is great because now patients only need to take one tablet,” stated Dr. Malhotra. AUGTYRO can be taken with or without food.1 Patients should be advised not to drink grapefruit juice or eat grapefruit while taking AUGTYRO.1 Capsules should be swallowed whole at approximately the same time every day as prescribed.1 Contents of the capsule should not be opened, crushed, chewed, or dissolved.1 If a dose is missed or if a patient vomits at any time after taking a dose, instruct patients to skip the dose and resume at a regularly scheduled time.1 Two doses should not be taken at the same time.1 Adjustable dosing allows for dose modification if needed for adverse reactions. Recommended dosage reductions for adverse reactions are the following1:
- For the dose of 160 mg QD:
- First dose reduction: 120 mg QD
- Second dose reduction: 80 mg QD
- For the dose of 160 mg BID:
- First dose reduction: 120 mg BID
- Second dose reduction: 80 mg BID
A prescription for 40-mg capsules is required for dose reductions.1 Additional detailed dose reduction recommendations are available for key adverse reactions.1
Summary and conclusions
AUGTYRO is the next-generation TKI helping patients with ROS1+ NSCLC start strong.1,2,9 Results from the TRIDENT-1 trial and continued response for TKI-naïve patients at the ~3-year follow-up analysis support its current place in therapy.1,2,9 “AUGTYRO is definitely my preferred drug for 1L ROS1+ NSCLC treatment—we are seeing responses for years,” stated Dr. Malhotra.
1L=first line; ATP=adenosine triphosphate; BICR=blinded independent central review; BID=twice daily; CI=confidence interval; CNS=central nervous system; cORR=confirmed ORR; CR=complete response; DOR=duration of response; ECOG PS=Eastern Cooperative Oncology Group performance status; EXP=expansion cohort; icORR=intracranial ORR; mDOR=median DOR; mNSCLC=metastatic NSCLC; NE=not evaluable; NSCLC=non-small cell lung cancer; ORR=overall response rate; PFS=progression-free survival; PR=partial response; QD=everyday; QTc=corrected QT; RECIST=Response Evaluation Criteria in Solid Tumors; ROS1=ROS proto oncogene 1; RP2D=recommended phase 2 dose; TKI=tyrosine kinase inhibitor.
INDICATION
AUGTYRO® (repotrectinib) is indicated for the treatment of adult patients with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC).
IMPORTANT SAFETY INFORMATION
Warnings & Precautions
Central Nervous System Adverse Reactions
• Among the 426 patients who received AUGTYRO in Study TRIDENT-1, a broad spectrum of central nervous system (CNS) adverse reactions including dizziness, ataxia, and cognitive disorders occurred in 77% of patients with Grade 3 or 4 events occurring in 4.5%.
• Dizziness, including vertigo, occurred in 65%; Grade 3 dizziness occurred in 2.8% of patients. The median time to onset was 7 days (1 day to 1.4 years). Dose interruption was required in 9% of patients, and 11% required dose reduction of AUGTYRO due to dizziness.
• Ataxia, including gait disturbance and balance disorder, occurred in 28% of patients; Grade 3 ataxia occurred in 0.5%. The median time to onset was 15 days (1 day to 1.4 years). Dose interruption was required in 5% of patients, 8% required dose reduction and one patient (0.2%) permanently discontinued AUGTYRO due to ataxia.
• Cognitive impairment, including memory impairment and disturbance in attention, occurred in 25% of patients. Cognitive impairment included memory impairment (15%), disturbance in attention (12%), and confusional state (2%); Grade 3 cognitive impairment occurred in 0.9% of patients. The median time to onset of cognitive disorders was 37 days (1 day to 1.4 years). Dose interruption was required in 2% of patients, 2.1% required dose reduction and 0.5% permanently discontinued AUGTYRO due to cognitive adverse reactions.
• Mood disorders occurred in 6% of patients. Mood disorders occurring in >1% of patients included anxiety (2.6%); Grade 4 mood disorders (mania) occurred in 0.2% of patients. Dose interruption was required in 0.2% of patients and 0.2% required a dose reduction due to mood disorders.
• Sleep disorders including insomnia and hypersomnia occurred in 18% of patients. Sleep disorders observed in >1% of patients were somnolence (9%), insomnia (6%) and hypersomnia (1.6%). Dose interruption was required in 0.7% of patients, and 0.2% required a dose reduction due to sleep disorders.
• The incidences of CNS adverse reactions reported were similar in patients with and without CNS metastases.
• Advise patients not to drive or use machines if they are experiencing CNS adverse reactions. Withhold and then resume at same or reduced dose upon improvement, or permanently discontinue AUGTYRO based on severity.
Interstitial Lung Disease (ILD)/Pneumonitis
• Among the 426 patients treated with AUGTYRO, ILD/pneumonitis (pneumonitis [2.8%] and ILD [0.2%]) occurred in 3.1%; Grade 3 ILD/pneumonitis occurred in 1.2%. The median time to onset was 45 days (19 days to 0.9 years). Dose interruption was required in 1.4% of patients, 0.5% required dose reduction, and 1.1% permanently discontinued AUGTYRO due to ILD/pneumonitis.
• Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold AUGTYRO in patients with suspected ILD/pneumonitis and permanently discontinue AUGTYRO if ILD/pneumonitis is confirmed.
Hepatotoxicity
• Among the 426 patients treated with AUGTYRO, increased alanine transaminase (ALT) occurred in 38%, increased aspartate aminotransferase (AST) occurred in 41%, including Grade 3 or 4 increased ALT in 3.3% and increased AST in 2.9%. The median time to onset of increased ALT or AST was 15 days (range: 1 day to 1.9 years). Increased ALT or AST leading to dose interruptions or reductions occurred in 2.8% and 1.2% of patients, respectively. Hyperbilirubinemia leading to dose interruptions occurred in 0.5%.
• Monitor liver function tests, including ALT, AST and bilirubin, every 2 weeks during the first month of treatment, then monthly thereafter and then as clinically indicated. Withhold and then resume at same or reduced dose upon improvement or permanently discontinue AUGTYRO based on the severity.
Myalgia with Creatine Phosphokinase (CPK) Elevation
• AUGTYRO can cause myalgia with or without creatine phosphokinase (CPK) elevation. Among the 426 patients treated with AUGTYRO, myalgia occurred in 13% of patients, with Grade 3 in 0.7%. Median time to onset of myalgia was 19 days (range: 1 day to 2 years). Concurrent increased CPK within a 7-day window was observed in 3.7% of patients. AUGTYRO was interrupted in one patient with myalgia and concurrent CPK elevation.
• Advise patients to report any unexplained muscle pain, tenderness, or weakness. Monitor serum CPK levels during AUGTYRO treatment and monitor CPK levels every 2 weeks during the first month of treatment and as needed in patients reporting unexplained muscle pain, tenderness, or weakness. Initiate supportive care as clinically indicated. Based on severity, withhold and then resume AUGTYRO at same or reduced dose upon improvement.
Hyperuricemia
• Among the 426 patients treated with AUGTYRO, 21 patients (5%) experienced hyperuricemia reported as an adverse reaction, 0.7% experienced Grade 3 or 4 hyperuricemia. One patient without pre-existing gout required urate-lowering medication.
• Monitor serum uric acid levels prior to initiating AUGTYRO and periodically during treatment. Initiate treatment with urate-lowering medications as clinically indicated. Withhold and then resume at same or reduced dose upon improvement, or permanently discontinue AUGTYRO based on severity.
Skeletal Fractures
• Among 426 adult patients who received AUGTYRO, fractures occurred in 2.3%. Fractures involved the ribs (0.5%), feet (0.5%), spine (0.2%), acetabulum (0.2%), sternum (0.2%), and ankles (0.2%). Some fractures occurred at sites of disease and prior radiation therapy. The median time to fracture was 71 days (range: 31 days to 1.4 years). AUGTYRO was interrupted in 0.3% of patients.
• Of 26 evaluable patients in an ongoing open-label study in pediatric patients, fractures occurred in one 12-year-old patient (ankle/foot) and one 10-year-old patient (stress fracture). AUGTYRO was interrupted in both patients. AUGTYRO is not approved for use in pediatric patients less than 12 years of age.
• Promptly evaluate patients with signs or symptoms (e.g., pain, changes in mobility, deformity) of fractures. There are no data on the effects of AUGTYRO on healing of known fractures and risk of future fractures.
Embryo-Fetal Toxicity
• Based on literature reports in humans with congenital mutations leading to changes in tropomyosin receptor tyrosine kinase (TRK) signaling, findings from animal studies, and its mechanism of action, AUGTYRO can cause fetal harm when administered to a pregnant woman.
• Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective non-hormonal contraception during treatment with AUGTYRO and for 2 months following the last dose, since AUGTYRO can render some hormonal contraceptives ineffective.
• Advise male patients with female partners of reproductive potential to use effective contraception during treatment with AUGTYRO and for 4 months after the last dose.
Adverse Reactions
• The safety of AUGTYRO was evaluated in 426 patients in TRIDENT-1. The most common adverse reactions (≥20%) were dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea.
Drug Interactions
Effects of Other Drugs on AUGTYRO
Strong and Moderate CYP3A Inhibitors
• Avoid concomitant use with strong or moderate CYP3A inhibitors. Concomitant use of AUGTYRO with a strong or a moderate CYP3A inhibitor may increase repotrectinib exposure, which may increase the incidence and severity of adverse reactions of AUGTYRO. Discontinue CYP3A inhibitors for 3 to 5 elimination half-lives of the CYP3A inhibitor prior to initiating AUGTYRO.
P-gp Inhibitors
• Avoid concomitant use with P-gp inhibitors. Concomitant use of AUGTYRO with a P-gp inhibitor may increase repotrectinib exposure, which may increase the incidence and severity of adverse reactions of AUGTYRO.
Strong and Moderate CYP3A Inducers
• Avoid concomitant use with strong or moderate CYP3A inducers. Concomitant use of AUGTYRO with a strong or moderate CYP3A inducer may decrease repotrectinib plasma concentrations, which may decrease efficacy of AUGTYRO.
Effects of AUGTYRO on other Drugs
Certain CYP3A4 Substrates
• Avoid concomitant use unless otherwise recommended in the Prescribing Information for CYP3A substrates, where minimal concentration changes can cause reduced efficacy. If concomitant use is unavoidable, increase the CYP3A4 substrate dosage in accordance with approved product labeling.
• Repotrectinib is a CYP3A4 inducer. Concomitant use of repotrectinib decreases the concentration of CYP3A4 substrates, which can reduce the efficacy of these substrates.
Contraceptives
• Repotrectinib is a CYP3A4 inducer, which can decrease progestin or estrogen exposure to an extent that could reduce the effectiveness of hormonal contraceptives.
• Avoid concomitant use of AUGTYRO with hormonal contraceptives. Advise females of childbearing potential to use an effective nonhormonal contraceptive.
Please see US Full Prescribing Information for AUGTYRO.
References:
1. AUGTYRO [package insert]. Princeton, NJ: Bristol-Myers Squibb Company.
2. Drilon A, Camidge DR, Lin JJ, et al. Repotrectinib in ROS1 fusion–positive non–small-cell lung cancer. N Engl J Med. 2024;390(2):118-131.
3. Lin JJ, Shaw AT. Recent advances in targeting ROS1 in lung cancer. J Thorac Oncol. 2017;12(11):1611-1625.
4. Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190-1203.
5. US Food and Drug Administration. FDA Approves Crizotinib Capsules. Published March 11, 2016. Accessed October 21, 2024. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizotinib-capsules.
6. Drilon A, Chiu CH, Fan Y, et al. Long-Term Efficacy and Safety of Entrectinib in ROS1 Fusion-Positive NSCLC. JTO Clin Res Rep. 2022;3(6):100332. Published 2022 Apr 29. doi:10.1016/j.jtocrr.2022.100332.
7. Shaw AT, Riely GJ, Bang YJ, et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol. 2019;30(7):1121-1126. doi:10.1093/annonc/mdz131.
8. US Food and Drug Administration. Center for Drug Evaluation and Research. AUGTYRO Label and Approval History. NDA218213. Published November 15, 2023. Accessed October 16, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/218213Orig1s000ltr.pdf.
9. Drilon A, Dziadziuszko R, Camidge DR, et al. Repotrectinib in tyrosine kinase inhibitor (TKI)-naïve patients with advanced ROS1 fusion-positive (ROS1+) NSCLC in the phase 1/2 TRIDENT-1 trial: clinical update, treatment beyond progression and subsequent therapies. Oral presentation at ASCO 2024. Poster 386.
10. Cho BC, Camidge DR, Lin JJ, et al. Repotrectinib in patients with ROS1 fusion-positive non-small cell lung cancer: update from the pivotal phase 1/2 TRIDENT-1 trial. Oral presentation at WCLC 2023. Abstract OA03.06.
11. ClinicalTrials.gov. A study of repotrectinib (TPX-0005) in patients with advanced solid tumors harboring ALK, ROS1, or NTRK1-3 rearrangements. Accessed April 19, 2024. https://clinicaltrials.gov/study/NCT03093116.
12. Cho BC, Lin JJ, Camidge DR, et al. Pivotal topline data from the phase 1/2 TRIDENT-1 trial of repotrectinib in patients with ROS1+ advanced non-small cell lung cancer (NSCLC). Oral presentation at ENA 2022. Abstract 2LBA.
13. Drilon A, Ou SI, Cho BC, et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 2018;8(10):1227-1236.
14. Murray BW, Rogers E, Zhai D, et al. Molecular Characteristics of Repotrectinib That Enable Potent Inhibition of TRK Fusion Proteins and Resistant Mutations. Mol Cancer Ther. 2021;20(12):2446-2456. doi:10.1158/1535-7163.MCT-21-0632
© 2024 Bristol-Myers Squibb Company. AUGTYRO®, is a registered trademark of
Bristol-Myers Squibb Company.
3600-US-2400322 11/24
