Written by: Debra Patt, MD, PhD, MBA
In our lifetime, the CDK 4/6 inhibitors have improved the quality of life and progression-free survival for patients with estrogen receptor (ER)-positive/human epidermal growth factor 2- (HER2)-negative breast cancer more than any other drug. Giving patients the opportunity for treatment allows them to realize the dream of modern cancer therapy. Over time, these drugs continue to show great promise in the metastatic setting and in high-risk adjuvant breast cancer patients. Understanding their optimal use and managing their toxicity will get us closer to supporting our patients to live well without cancer. This article will address abemaciclib in metastatic breast cancer and also its use in early-stage breast cancer, including the update of FDA guidance and also data including 4-year follow up.
Abemaciclib in Metastatic Breast Cancer
The first CDK4/6 inhibitor palbociclib, was approved by the FDA in 2016, followed by ribociclib and abemaciclib which were approved the following year. These drugs as a class have made a palpable difference in the lives of breast cancer patients. They have not only improved progression-free and overall survival but have also allowed patients with advanced cancer to live with the disease without the burden of highly toxic intravenous chemotherapy. In that way, many patients control their cancer just like hypertension or other chronic illnesses, with pills that have minimal impact on their quality of life.
The three CDK4/6 inhibitors are often discussed comparatively, but we do not yet have direct comparative data, limiting decisions on therapy to our understanding of each of them individually and their efficacy and toxicity profiles.
Some differences of importance across the drugs in the metastatic setting are efficacy and toxicity. See Table 1 for the designs of the metastatic trials and their efficacy in comparison to the control arms. In addition, there are important differences in adverse effect profiles, seen in Table 2. It is notable that in the frontline trials, many patients were managed with dose reduction. This is an important point that will be touched upon again and again, that there is no compelling evidence that efficacy is sacrificed when dose reduction is managed to abate toxicity. More specifically, given the absence of data on dose response curves and the high rates of discontinuation due to toxicity, practitioners should be eager to manage symptoms with supportive care medications and dose reduction. Specifically, when we initiate patients on treatment with abemaciclib, they should be followed closely—initially, weekly or every other week—and dose should be reduced rapidly as indicated to manage symptoms. Similarly empowering patients with education and administering anti-diarrheal therapy to manage toxicity with initial prescribing can go a long way to assist in symptom control. Taking these actions up front could prevent early discontinuation of effective therapy.
Table 1: Summary data of efficacy of frontline CDK4/6 inhibitors in postmenopausal ER-positive breast cancer patients.
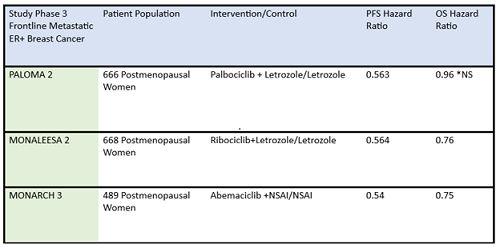
ER+, estrogen receptor positive; NS, not significant; NSAI, nonsteroidal aromatase inhibitor; OS, overall survival; PFS, progression-free survival
*Paloma 2 hazard ratio for OS was not statistically significant
Table 2: Summary of adverse events (AE) and serious adverse events (SAE) of frontline CDK4/6 inhibitors in post-menopausal ER-positive breast cancer patients
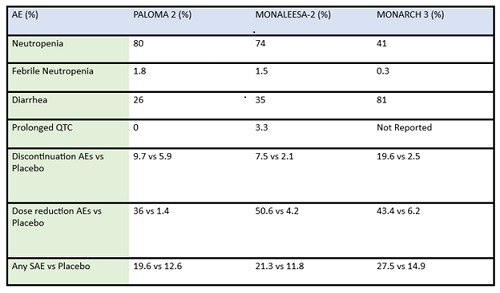
There are some key differences in how CDK4/6 inhibitors are used in the metastatic setting: activity in combination vs as a single agent, penetration of the blood brain barrier, and evidence for benefit from treatment after progressing on another drug in the same class. For example, abemaciclib is FDA approved as a single agent showing activity with doses at 200mg every 12 hours for patients with metastatic ER-positve/HER2-negative breast cancer1. Abemaciclib has activity in the central nervous system, and is included in the ASCO guidelines among the active agents in ER-positive/HER2-amplified breast cancer with brain metastasis2. Abemaciclib may be an effective therapy after treatment with palbociclib, as a recent cohort of 52 patients previously treated with palbociclib exhibited a clinically meaningful benefit from subsequent therapy with abemaciclib3.
Abemaciclib in Adjuvant High-Risk ER-Positive/HER2-Negative Breast Cancer
Observing the success in patients with metastatic breast cancer, we are seeking to understand if treatment is beneficial in earlier lines of therapy. The MONARCH E trial, evaluating the efficacy and safety of abemaciclib in combination with endocrine blockade in patients with node-positive high-risk ER-positive breast cancer, demonstrated an improvement in disease-free survival. This has been a clinically meaningful addition to our armamentarium of treatment, although careful consideration of management is important as early failure to manage adverse effects can lead to early discontinuation. According to the 4-year follow-up data from MONARCH E, the median invasive disease-free survival benefit previously reported of HR=0.664 (95% CI 0.578-0.762, nominal p<0.0001) was persistent and the absolute difference in invasive disease-free survival was 6.4% (85.8% in the endocrine therapy plus abemaciclib arm versus 79.4% in the endocrine only arm). Overall survival did not meet statistical significance, and the adverse effect profile reflected toxicities known to be associated with abemaciclib, including neutropenia, leukopenia, and diarrhea4. Adjuvant abemaciclib was approved by the FDA in 2021 and is currently approved in combination with endocrine therapy (tamoxifen or an aromatase inhibitor) for the adjuvant treatment of adult patients with HR-positive, HER2-negative, node-positive early breast cancer at high risk of recurrence. Of note, in March 2023, the FDA approval was expanded to remove Ki-67 >20% as a qualifying factor for approval. Patients defined as high risk included those having ≥4 pathologically involved axillary lymph nodes or 1-3 axillary lymph nodes and either tumor grade 3 or tumor size >5cm.
Abemaciclib causes GI toxicity in the form of cramping and diarrhea. Frequently, patients are afflicted with this toxicity, and if they are not optimally managed with anti-diarrheal agents and dose reductions, the patients will prematurely discontinue effective therapy. This is a particular problem in the adjuvant patients: they have often already completed systemic chemotherapy, and their therapeutic enthusiasm wanes as they have completed what they often (incorrectly) perceive as the more important part of therapy. Critical attention to symptom management, patient education, and dose reduction are important, as prescribing at the FDA approved dose will sometimes cause intolerable adverse effects, and early dose reduction will likely lead to reduction of adverse effects and improved compliance with the adjuvant treatment strategy. With all of the CDK4/6 inhibitors there is a large amount of inter-individual variability in exposure, yet in contrast to palbociclib and ribociclib, abemaciclib has three active metabolites that all have clinical activity5. As we don’t have a robust amount of clinical data on dose response to abemaciclib, there has been some hesitation among practitioners to implement strategies to manage toxicity early with dose reduction. Anecdotally, some strategies that have been effective in managing adverse effects include giving a smaller allocation of the drug and seeing the patient 1 and 2 weeks out in follow up, quickly reducing the dose, and sometimes even starting at a lower dose initially. In addition, partnering a new therapy regimen with patient education and loperamide to manage adverse effects can assist in helping patients avoid and manage severe toxicity.
The biggest challenge I have anecdotally observed in clinical practice in patients benefitting from adjuvant abemaciclib is that qualifying patients often don’t have it prescribed for them as part of their adjuvant therapy. Adjuvant abemaciclib was approved in 2021 by the FDA, and while adoption does take time, adoption in clinical practice has been variable.
Clinical Take Aways: When prescribing abemaciclib in patients with metastatic breast cancer, patient education, up-front management of diarrhea, and close follow-up for dose modification and symptom management needs are critical. When prescribing abemaciclib in patients with high-risk ER-positive HER2-negative breast cancer, education, close follow-up, dose modification, and prescribing loperamide to accompany the therapy are also important. Above all, be sure to discuss with high-risk patients the opportunity to reduce their risk with appropriate therapy and the importance of therapy adherence in achieving favorable outcomes.
References
1) Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, Wildiers H, Hudis CA, O’Shaughnessy J, Zamora E, Yardley DA, Frenzel M, Koustenis A, Baselga J. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2- Metastatic Breast Cancer. Clin Cancer Res. 2017 Sep 1;23(17):5218-5224. doi: 10.1158/1078-0432.CCR-17-0754. Epub 2017 May 22. Erratum in: Clin Cancer Res. 2018 Nov 1;24(21):5485. PMID: 28533223; PMCID: PMC5581697.
2) Giordano SH, Franzoi MAB, Temin S, Anders CK, Chandarlapaty S, Crews JR, Kirshner JJ, Krop IE, Lin NU, Morikawa A, Patt DA, Perlmutter J, Ramakrishna N, Davidson NE. Systemic Therapy for Advanced Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: ASCO Guideline Update. J Clin Oncol. 2022 Aug 10;40(23):2612-2635. doi: 10.1200/JCO.22.00519. Epub 2022 May 31. PMID: 35640077.
3) Navarro-Yepes J, Kettner NM, Rao X, Bishop CS, Bui TN, Wingate HF, Singareeka Raghavendra A, Wang Y, Wang J, Sahin AA, Meric-Bernstam F, Hunt KK, Damodaran S, Tripathy D, Keyomarsi K. Abemaciclib is effective in palbociclib-resistant hormone receptor-positive metastatic breast cancers. Cancer Res. 2023 Jun 29:CAN-23-0705. doi: 10.1158/0008-5472.CAN-23-0705. Epub ahead of print. PMID: 37384539.
4) Johnston SRD, Toi M, O’Shaughnessy J, Rastogi P, Campone M, Neven P, Huang CS, Huober J, Jaliffe GG, Cicin I, Tolaney SM, Goetz MP, Rugo HS, Senkus E, Testa L, Del Mastro L, Shimizu C, Wei R, Shahir A, Munoz M, San Antonio B, André V, Harbeck N, Martin M; monarchE Committee Members. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 2023 Jan;24(1):77-90. doi: 10.1016/S1470-2045(22)00694-5. Epub 2022 Dec 6. PMID: 36493792.
5) Groenland SL, Martínez-Chávez A, van Dongen MGJ, Beijnen JH, Schinkel AH, Huitema ADR, Steeghs N. Clinical Pharmacokinetics and Pharmacodynamics of the Cyclin-Dependent Kinase 4 and 6 Inhibitors Palbociclib, Ribociclib, and Abemaciclib. Clin Pharmacokinet. 2020 Dec;59(12):1501-1520. doi: 10.1007/s40262-020-00930-x. PMID: 33029704.
