SUMMARY: According to the American Cancer Society, tobacco use is responsible for about 1 in 5 deaths in the United States and is the leading preventable cause of death in the US. Smoking (cigarettes, cigars, and pipes) is responsible for about 20% of all cancers and about 30% of all cancer deaths in the US. Approximately 80% of lung cancers, as well as about 80% of all lung cancer deaths, are due to smoking, and lung cancer is the leading cause of cancer death in both men and women. Smoking also increases the risk for cancers of the Oral cavity, Oropharynx, Larynx, Esophagus, Stomach, Liver, Pancreas, Colon/Rectum, Kidney, Bladder, Cervix, as well as Acute Myeloid Leukemia. The American Cancer Society estimates that for 2022, about 236,740 new cases of lung cancer will be diagnosed and 135,360 patients will die of the disease. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
Previous published studies have shown that individuals who start smoking at a younger age have greater mortality risk than those who start smoking later in life, and smoking cessation especially at younger ages substantially reduces that mortality risk. Several biologic mechanisms have been hypothesized, to explain the beneficial effect of smoking cessation on survival, in patients with Lung Cancer. Tobacco smoke has been shown to promote tumor growth, progression, and dissemination, while decreasing the efficacy and tolerance to radiation and systemic therapy. Further, it is well established that smoking increases the risk of postoperative complications and second primary cancers. Epigenetic changes induced by tobacco smoke may play an important role, and cigarette smoke induced diseases may be the result of alterations in DNA methylation, and is a reversible gene regulatory modification. Following smoking cessation, the majority of the differentially methylated CpG dinucleotide sites of the current smokers return to the level of the never smokers within 5 years of smoking cessation. However, some of the methylated genes may not return to the level of the never smokers even after 30 years of smoking cessation, suggesting that tobacco smoke can lead to lasting damage to human health. In this publication, the researchers aimed to summarize the current scientific evidence on whether quitting smoking at or around diagnosis has a beneficial effect on the survival of Lung Cancer patients.
The authors conducted a systematic literature review and meta-analysis of the studies that evaluated the prognostic effect of quitting smoking at or around diagnosis among patients with Lung Cancer. The meta-analysis included 21 articles published between 1980 and October 2021 on the effect of smoking cessation at or around the time of diagnosis among a total of 10,938 patients with lung cancer, which included patients with Non Small Cell Lung Cancer, Small Cell Lung Cancer, as well as patients with Lung Cancer of both or unspecified subtypes or whose subtype was not specified. In most studies analyzed, the median age at Lung Cancer diagnosis was between 60 and 70 years. The authors used random effect meta-analysis models to pool study-specific data into Summary Relative Risk (SRR) and corresponding 95% confidence intervals (CI). There was substantial variability across studies with regards to study design, patient characteristics, treatments received, criteria used to define smoking status (quitters or continued), and duration of follow up.
Even though there was moderate heterogeneity of Hazard Ratio across studies, it was noted that quitting smoking at or around diagnosis was associated with a significant 29% improvement in Overall Survival, compared with patients who continued to smoke after their diagnosis (SRR=0.71; 95% CI 0.64–0.80). This benefit of quitting smoking was noted regardless of lung cancer histologic subtype, with a 20-30% reduction in the risk of death among those who quit smoking post-diagnosis, compared to those who continued to smoke.
It was concluded from this analysis that quitting smoking at or around diagnosis is associated with a beneficial effect on the survival of Lung Cancer patients, and smoking cessation can be nearly as effective in improving the chance of survival as treatment with chemotherapy, immunotherapy or radiation therapy. The authors added that based on these finding, Health Care Providers should educate Lung Cancer patients about the benefits of quitting smoking even after diagnosis and provide them with the necessary support for smoking cessation.
Quitting Smoking At or Around Diagnosis Improves the Overall Survival of Lung Cancer Patients: A Systematic Review and Meta-Analysis. Caini S, Riccio MD, Vettori V, et al. Published:January 04, 2022. DOI:https://doi.org/10.1016/j.jtho.2021.12.005

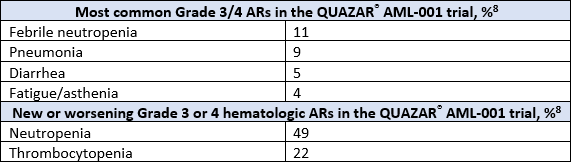
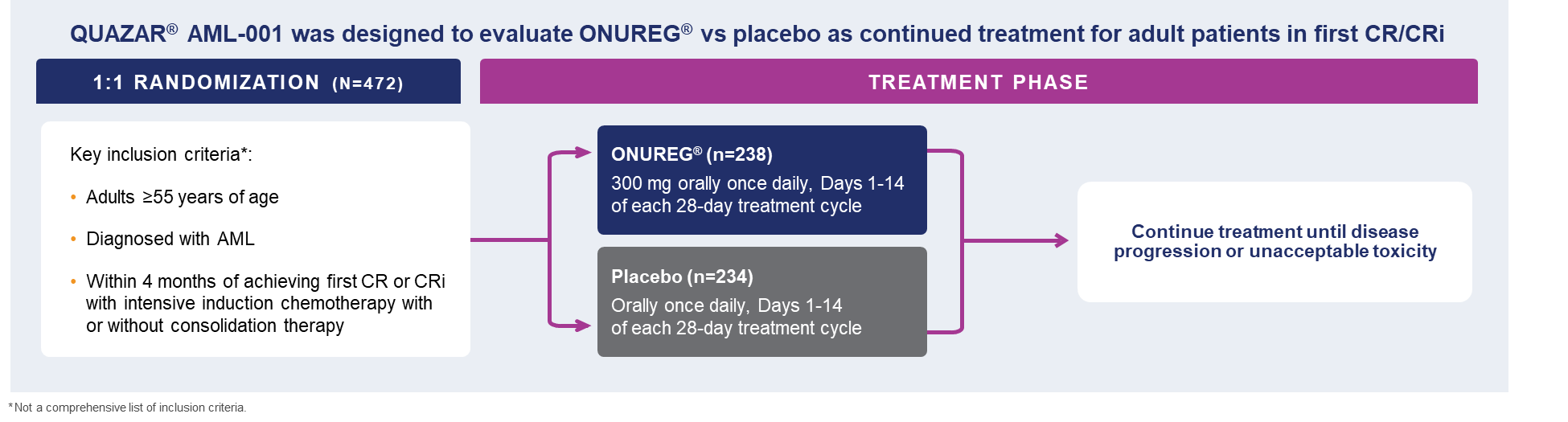 A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8
A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8