SUMMARY: The American Cancer Society estimates that for 2021, about 21,250 new cases of CLL will be diagnosed in the US and 4320 patients will die of the disease. CLL accounts for about one-quarter of the new cases of leukemia. The average age of patients diagnosed with CLL is around 70 years, and is rarely seen in people under age 40, and is extremely rare in children.
The pro-survival (anti-apoptotic) protein BCL2 is over expressed by CLL cells and regulates clonal selection and cell survival. A new class of anticancer agents known as BH3-mimetic drugs mimic the activity of the physiologic antagonists of BCL2 and related proteins and promote apoptosis (programmed cell death). VENCLEXTA® is a second generation, oral, selective, small molecule inhibitor of BCL2 and restores the apoptotic processes in tumor cells.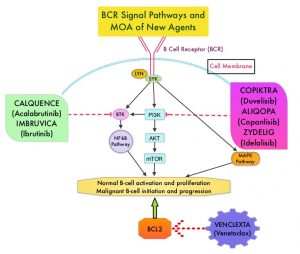
CLL14 Trial is a prospective, multicenter, open-label, randomized Phase III study conducted in close collaboration with the German CLL Study Group (DCLLSG). This study was designed to evaluate the efficacy and safety of a fixed duration combination of VENCLEXTA® and GAZYVA® (Obinutuzumab) versus GAZYVA® and Chlorambucil in previously-untreated patients with CLL and coexisting medical conditions. In this trial, 432 treatment-naïve patients with CLL were randomized in a 1:1 ratio to receive fixed duration of 12 months of VENCLEXTA® in combination with six cycles of GAZYVA®, or 6 cycles of GAZYVA® in combination with Chlorambucil. Both treatment groups were well balanced and the median patient age was 72 years. The Primary endpoint was Progression Free Survival (PFS) assessed by an Independent Review Committee. Secondary endpoints included Minimal Residual Disease (MRD) status, Overall Response Rate, Complete Response, Complete Remission with Incomplete Hematologic Recovery (CRi), Overall Survival, duration of response, Time to Next CLL Treatment, and safety.
The median PFS was not reached in either treatment groups after a median follow-up of 28 months. The trial demonstrated a statistically significant improvement in PFS for patients who received VENCLEXTA® plus GAZYVA®, compared with those who received GAZYVA® plus Chlorambucil (HR 0.33; P<0.0001), suggesting a 67% reduction in the risk of progression or death with the VENCLEXTA® plus GAZYVA® combination. The Overall Response Rate was 85% in VENCLEXTA® plus GAZYVA® group compared to 71% in GAZYVA® plus Chlorambucil group (P=0.0007). Based on this data, the FDA in May 2019 approved VENCLEXTA® (Venetoclax) as frontline treatment for adult patients with Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma (SLL).
The authors in this presentation provided updated efficacy and safety data from the ongoing follow up of the CLL14 study, with all patients off study treatment for at least 3 years. After a median follow-up of 52.4 months, PFS continued to be superior for VENCLEXTA® plus GAZYVA® group, compared to GAZYVA® plus Chlorambucil (median Not Reached versus 36.4 months; HR=0.33 P<0.0001). At 4 years after randomization, the estimated PFS rate was 74.0% in the VENCLEXTA® plus GAZYVA® arm and 35.4% in the GAZYVA® plus Chlorambucil arm. This benefit was noted across all clinical and biological risk groups, including patients with TP53 mutation/deletion (4-year PFS 53.0% versus 20.8%) and unmutated IGHV status (4-year PFS 68.0% versus 19.8%). Time to Next Treatment was significantly longer in the VENCLEXTA® plus GAZYVA® group, compared to GAZYVA® plus Chlorambucil group (4-year TTNT 81.1% versus 59.9%; HR=0.46, P<0.0001). Further, majority of patients received and responded to BTK inhibitor monotherapy as a second-line treatment after progressive disease in both the treatment groups.
Assessment of MRD in peripheral blood 30 months after the end of treatment showed that 26.9% of patients in the VENCLEXTA® group still had undetectable MRD (less than 10-4), compared with 3.2% in the GAZYVA® plus Chlorambucil group. The median OS has not yet been reached in either treatment groups. No new safety signals were observed.
It was concluded that the fixed duration combination of VENCLEXTA® plus GAZYVA® continued to confer a PFS advantage over GAZYVA® plus Chlorambucil, for patients with previously untreated CLL, and remains an effective treatment for all patients with CLL and with coexisting conditions.
Venetoclax-obinutuzumab for previously untreated chronic lymphocytic leukemia: 4-year follow-up analysis of the randomized CLL14 study. Al-Sawaf O, Zhang C, Robrecht S, et al. Presented at: European Hematology Association 2021 Virtual Congress; June 9-17, 2021. Abstract S146.