SUMMARY: Prostate cancer is the most common cancer in American men with the exclusion of skin cancer, and 1 in 9 men will be diagnosed with prostate cancer during their lifetime. It is estimated that in the United States, about 191,930 new cases of prostate cancer will be diagnosed in 2020 and 33,330 men will die of the disease. The development and progression of prostate cancer is driven by androgens. Androgen Deprivation Therapy (ADT) or testosterone suppression has therefore been the cornerstone of treatment of advanced prostate cancer, and is the first treatment intervention. Androgen Deprivation Therapies have included bilateral orchiectomy or Gonadotropin Releasing Hormone (GnRH) analogues, with or without first generation Androgen Receptor (AR) inhibitors such as CASODEX® (Bicalutamide), NILANDRON® (Nilutamide) and EULEXIN® (Flutamide) or with second-generation, anti-androgen agents, which include ZYTIGA® (Abiraterone), XTANDI® (Enzalutamide), ERLEADA® (Apalutamide) and NUBEQA® (Darolutamide).
Androgen Deprivation Therapies such as GnRH analogs/Luteinizing Hormone Releasing Hormone (LHRH) agonists are standard treatment for patients with advanced prostate cancer. These agents when first administered cause an initial surge in Luteinizing Hormone, Follicle Stimulating Hormone (FSH), and testosterone levels. With continuous administration, LHRH agonists desensitize the pituitary receptor and suppress the production of Luteinizing Hormone and testosterone, thus blocking the pulsatile secretion of GnRH by the hypothalamus. LHRH agonists however do not fully suppress FSH which is a potential mitogenic growth factor for prostate cancer cells. The initial testosterone surge may result in flaring up of symptoms such as bone pain, obstructive urinary symptoms, and rarely spinal cord compression. For this reason, anti-androgen agents are recommended for the first few weeks after initiation of an LHRH agonist. LHRH agonists have been shown to increase the near-term risk of major adverse cardiovascular events, by promoting plaque destabilization and rupture.
Degarelix (FIRMAGON®) is a GnRH antagonist, and the depot injection was approved by the FDA in December 2018. Degarelix suppresses both Luteinizing Hormone and FSH, resulting in rapid testosterone suppression, without an initial testosterone surge. This agent however has to be administered monthly and approximately 40% of patients experience reactions at the injection site.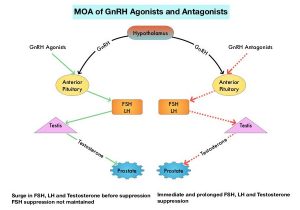
Relugolix is a highly selective, GnRH antagonist that can be given orally once daily, and has a half-life of 25 hours. In multiple Phase I and Phase II studies, Relugolix has been shown to lower testosterone levels by rapidly inhibiting the pituitary release of Luteinizing Hormone and FSH. The HERO trial is a multinational, randomized, open-label, Phase III study, which evaluated the efficacy and safety of Relugolix, an oral GnRH antagonist, as compared with those of Leuprolide (LUPRON®) (GnRH agonist), in men with advanced prostate cancer. In this study, a total of 930 patients were randomly assigned in a 2:1 ratio to receive either Relugolix 120 mg orally once daily, after a single oral loading dose of 360 mg (N=622) or Leuprolide acetate 22.5 mg IM every 3 months (N=308), for 48 weeks.
Eligible patients had one of three clinical disease presentations: 1) Evidence of biochemical (PSA) or clinical relapse after local primary intervention with curative intent 2) Newly diagnosed hormone-sensitive metastatic disease, or 3) Advanced localized disease unlikely to be cured by local primary intervention with curative intent. Patients with major adverse cardiovascular events within 6 months before trial initiation were excluded. Patients were stratified according to the presence or absence of metastatic disease, and age (75 yrs or less and over 75 years). Approximately 32% of patients had metastatic disease and 50% had biochemical recurrence after definitive treatment.
The Primary endpoint was sustained testosterone suppression to castrate levels (less than 50 ng/dL) through 48 weeks. Secondary end points included noninferiority of Relugolix to Leuprolide with respect to sustained castration rate, castrate levels of testosterone on day 4, and profound castrate levels (less than 20 ng/dL) on day 15. Testosterone recovery after discontinuation of the trial drug was to be evaluated in a subgroup of patients. The median follow up time in both groups, including the 30-day safety follow-up period for adverse events, was 52 weeks.
Relugolix was associated with a significantly higher rate of maintained castrate levels of testosterone, when compared to Leuprolide. Castrate levels of testosterone were maintained through 48 weeks in 96.7% of patients in the Relugolix group compared to 88.8% of patients in the Leuprolide group. The difference of 7.9 percentage points showed noninferiority as well as superiority of Relugolix (P<0.001 for superiority) over Leuprolide. All other key Secondary end points showed superiority of Relugolix over Leuprolide (P<0.001). These endpoints included the percentage of patients with castrate levels of testosterone on day 4 (56% versus 0%) and on day 15 (98.7% versus 12%), testosterone suppression to less than 20ng/dL on day 15 (78.4% versus 1%) and confirmed PSA response of more than 50% decrease at day 15 (79.4% versus 19.8%; P<0.001). In the subgroup of 184 patients followed for testosterone recovery, the mean testosterone levels 90 days after treatment discontinuation were 288.4 ng/dL in the Relugolix group and 58.6 ng/dL in the Leuprolide group. The incidence of major adverse cardiovascular events among all the patients was 2.9% in the Relugolix group and 6.2% in the Leuprolide group (HR=0.46).
The authors concluded that in this trial involving men with advanced prostate cancer, Relugolix achieved rapid and sustained suppression of testosterone levels, that was superior to that with Leuprolide, with a 54% lower risk of major adverse cardiovascular events.
Oral Relugolix for Androgen-Deprivation Therapy in Advanced Prostate Cancer. Shore ND, Saad F, Cookson MS, et al. for the HERO Study Investigators. N Engl J Med 2020; 382:2187-2196.