SUMMARY: The FDA on April 29, 2020 approved ZEJULA® (Niraparib) for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in a complete or partial response to first-line platinum-based chemotherapy. It is estimated that in the United States, approximately 21,750 women will be diagnosed with ovarian cancer in 2020 and 13,940 women will die of the disease. Ovarian cancer ranks fifth in cancer deaths among women, and accounts for more deaths than any other cancer of the female reproductive system. Approximately 75% of the ovarian cancer patients are diagnosed with advanced disease. Patients with newly diagnosed advanced ovarian cancer are often treated with platinum based chemotherapy following primary surgical cytoreduction. Approximately 70% of these patients will relapse within the subsequent 3 years and are incurable, with a 5 year Overall Survival rate of about 20-30%.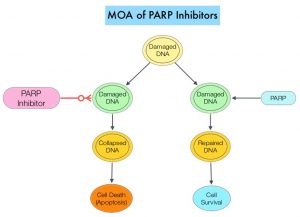
DNA damage is a common occurrence in daily life by UV light, ionizing radiation, replication errors, chemical agents, etc. This can result in single and double strand breaks in the DNA structure which must be repaired for cell survival. The two vital pathways for DNA repair in a normal cell are BRCA1/BRCA2 and PARP. The PARP (Poly ADP Ribose Polymerase) family of enzymes, include PARP1 and PARP2. In the context of DNA repair, BRCA1 and BRCA2 genes recognize and repair double strand DNA breaks via Homologous Recombination (HR) pathway. Homologous Recombination is a type of genetic recombination, and is a DNA repair pathway utilized by cells to accurately repair DNA double-stranded breaks during the S and G2 phases of the cell cycle, and thereby maintain genomic integrity. Homologous Recombination Deficiency (HRD) is noted following mutation of genes involved in HR repair pathway. At least 15 genes are involved in the Homologous Recombination Repair (HRR) pathway including BRCA1 and BRCA2 genes. The BRCA1 gene is located on the long (q) arm of chromosome 17 whereas BRCA2 is located on the long arm of chromosome 13. BRCA1 and BRCA2 are tumor suppressor genes and functional BRCA proteins repair damaged DNA, and play an important role in maintaining cellular genetic integrity. They regulate cell growth and prevent abnormal cell division and development of malignancy. Mutations in BRCA1 and BRCA2 account for about 20-25% of hereditary breast cancers and about 5-10% of all breast cancers. They also account for 15% of ovarian cancers, in addition to other cancers such as Colon and Prostate. BRCA mutations can either be inherited (Germline) and present in all individual cells or can be acquired and occur exclusively in the tumor cells (Somatic). Somatic mutations account for a significant portion of overall BRCA1 and BRCA2 aberrations. Loss of BRCA function due to frequent somatic aberrations likely deregulates HR pathway, and other pathways then come in to play, which are less precise and error prone, resulting in the accumulation of additional mutations and chromosomal instability in the cell, with subsequent malignant transformation. HRD therefore indicates an important loss of DNA repair function. Hereditary Epithelial Ovarian Cancer was thought to be caused almost exclusively by mutations in BRCA1 and BRCA2. It however is now well known that about 50% of the high grade serous ovarian cancers have aberrations in HR repair pathway. Deregulated HR pathway increases sensitivity to platinum drugs. Majority of the women with germline BRCA mutations (gBRCA) are positive for HR deficiency.
PARP is a related enzymatic pathway that repairs single strand breaks in DNA. In a BRCA mutant, the cancer cell relies solely on PARP pathway for DNA repair. In the presence of a PARP inhibitor, there is synthetic lethality because loss of both genes, leading to cell death. Thus PARP inhibitors are only harmful to cancer cells. ZEJULA® is a highly selective PARP 1/2 inhibitor, that causes cumulative DNA damage and cell death by inhibiting PARP. Previously published phase III study among patients with platinum-sensitive, recurrent ovarian cancer (NEJM 2016;375:2154-2164) concluded that Niraparib significantly prolonged Progression Free Survival (PFS) compared to placebo, and this benefit was achieved regardless of the presence or absence of germline BRCA mutations or HRD status.
PRIMA trial is a randomized, double-blind, placebo-controlled, international Phase III trial conducted to test the efficacy and safety of ZEJULA® maintenance therapy after a response to platinum-based chemotherapy, in patients with newly diagnosed advanced ovarian cancer at high risk for relapse. It should be noted that at the time PRIMA trial was designed, AVASTIN® (Bevacizumab) was not approved for first-line treatment in all participating countries. A total of 733 patients with newly diagnosed, high risk, advanced ovarian cancer were randomly assigned in a 2:1 ratio to receive ZEJULA® (N=487) or placebo (N=246) once daily in 28-day cycles for 36 months or until disease progression, after a response to platinum-based chemotherapy regimen. Patients received a dose of 200-300mg once daily, based on body weight and platelet count. Enrolled patients were at high risk for progressive disease with 23.1% having Stage III ovarian cancer with residual disease after primary debulking surgery, 66.7% had received neoadjuvant chemotherapy, 35% had Stage IV ovarian cancer, and 30.5% had a Partial Response to first-line platinum-based chemotherapy. Tumor samples were tested for HRD status and HRD was defined by either presence of tumor BRCA mutation or Genomic Instability Score (GIS) of 42 or more. Of the randomized patients, 50.9% had tumors with HRD, 30.4% had BRCA mutations and 20.5% were BRCA wild type. The treatment groups were well balanced. The Primary endpoint was Progression Free Survival (PFS) in patients who had tumors with HRD, and then in the overall population, as determined on hierarchical testing. Secondary end points included Overall Survival, time until the first subsequent therapy, PFS 2, defined as time from randomization to progression while the patient was receiving a subsequent anticancer therapy and Patient-Reported Outcomes. The median duration of follow-up at the time of the data cutoff was 13.8 months.
There was a statistically significant improvement in PFS for patients randomized to ZEJULA® compared with placebo in the HRD group, as well as the overall population. The median PFS in the HRD group was 21.9 months for patients receiving ZEJULA® compared with 10.4 months for those receiving placebo (HR=0.43; P<0.001). The median PFS in the overall population was 13.8 months for patients receiving ZEJULA® compared with 8.2 months for those receiving placebo (HR=0.62; P<0.001). At the 24-month interim analysis, the rate of Overall Survival was 84% in the ZEJULA® group and 77% in the placebo group (HR=0.70). The most common adverse reactions in patients receiving ZEJULA® were cytopenias, fatigue, AST/ALT elevation, hypertension, low grade nausea and decreased appetite.
It was concluded that among patients with newly diagnosed advanced ovarian cancer who had responded to platinum-based chemotherapy, ZEJULA® significantly prolonged Progression Free Survival, compared to those who received placebo, regardless of the presence or absence of Homologous Recombination Deficiency.
Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. González-Martín A, Pothuri B, Vergote I, et al. for the PRIMA/ENGOT-OV26/GOG-3012 Investigators. N Engl J Med 2019; 381:2391-2402