SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Approximately 15% of patients present with early stage (T1-2 N0) disease, and these numbers are likely to increase with the implementation of Lung Cancer screening programs. Patients with early stage disease unless medically unfit, undergo surgical resection with a curative intent. Those who are not surgical candidates, are often treated with conventional Radiation Therapy, which can result in high rates of local failure and treatment-related toxicities.
Stereotactic Body Radiation Therapy (SBRT) is a non-surgical procedure that allows delivery of significantly higher doses of precisely focused radiation to the tumor, compared to conventional Radiation Therapy, with less collateral damage to the surrounding normal tissue. The technologies used for SBRT include GAMMA KNIFE® which uses highly focused gamma rays, Proton Beam therapy which uses ionized hydrogen or Protons, Linear Accelerator (LINAC) and CYBER KNIFE® which use Photons, to target the tumor tissue. Because SBRT is fractionated and delivered over 1-5 days, the short-and long-term side effects of radiation therapy are decreased and may allow higher total dosage to be given.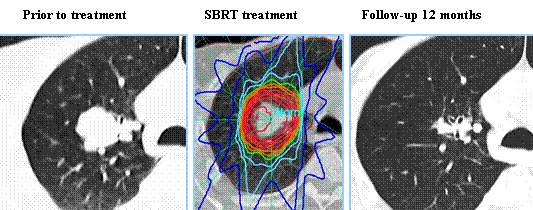
This guideline is based on systematic review of literature which included 172 articles, from January 1995 and August, 2016. This literature search evaluated adults with T1-2, N0, Non Small Cell Lung Cancer (NSCLC) receiving primary or salvage SBRT. Developed by the American Society for Radiation Oncology, this guideline is also endorsed by the European Society for Radiotherapy & Oncology, the Royal Australian and New Zealand College of Radiologists, and the International Association for the Study of Lung Cancer.
KEY QUESTIONS (KQ)
KQ 1: When is SBRT appropriate for patients with T1-2, N0, NSCLC who are medically operable?
Statement KQ 1A: Any patient with operable Stage I NSCLC being considered for SBRT should be evaluated by a thoracic surgeon, preferably in a multidisciplinary setting, to reduce specialty bias.
Statement KQ 1B: For patients with “standard operative risk” (ie, with anticipated operative mortality of <1.5%) and stage I NSCLC, SBRT is not recommended as an alternative to surgery outside of a clinical trial. Discussions about SBRT are appropriate, with the disclosure that long-term outcomes with SBRT >3 years are not well established. For this population, lobectomy with systematic mediastinal lymph node evaluation remains the recommended treatment, though a sublobar resection may be considered in select clinical scenarios.
Statement KQ 1C: For patients with “high operative risk” (ie, those who cannot tolerate lobectomy, but are candidates for sublobar resection) stage I NSCLC, discussions about SBRT as a potential alternative to surgery are encouraged. Patients should be informed that while SBRT may have decreased risks from treatment in the short term, the longer term outcomes >3 years are not well-established.
KQ 2: When is SBRT appropriate for medically inoperable patients with T1-2, N0, NSCLC?
For patients with centrally located tumors
Statement KQ 2A: SBRT directed toward centrally located lung tumors (tumor within 2 cm of the proximal tracheobronchial tree) carries unique and significant risks when compared to treatment directed at peripherally located tumors. The use of 3-fraction regimens should be avoided in this setting.
Statement KQ 2B: SBRT directed at central lung tumors should be delivered in 4 or 5 fractions. Adherence to volumetric and maximum dose constraints may optimize the safety profile of this treatment. For central tumors for which SBRT is deemed too high risk, hypofractionated radiation therapy utilizing 6 to 15 fractions can be considered.
For patients with tumors >5 cm in diameter
Statement KQ 2C: SBRT is an appropriate option for tumors >5 cm in diameter with an acceptable therapeutic ratio. Adherence to volumetric and maximum dose constraints may optimize the safety profile of this treatment.
For patients lacking tissue confirmation
Statement KQ 2D: Whenever possible, obtain a biopsy prior to treatment with SBRT to confirm a histologic diagnosis of a malignant lung nodule.
Statement KQ 2E: SBRT can be delivered in patients who refuse a biopsy, have undergone non-diagnostic biopsy, or who are thought to be at prohibitive risk of biopsy. Prior to SBRT in patients lacking tissue confirmation of malignancy, patients are recommended to be discussed in a multidisciplinary manner with a consensus that the lesion is radiographically and clinically consistent with a malignant lung lesion based on tumor, patient, and environmental factors
For patients with synchronous primary or multifocal tumors
Statement KQ 2F: Multiple Primary Lung Cancers (MPLCs) can be difficult to differentiate from intrathoracic metastatic lung cancer and pose unique issues for parenchymal preservation; therefore, it is recommended that they are evaluated by a multidisciplinary team.
Statement KQ 2G: Positron Emission Tomography/Computed Tomography and brain Magnetic Resonance Imaging are recommended in patients suspected of having MPLC to help differentiate from intrathoracic metastatic lung cancer. Invasive mediastinal staging should be addressed on a case-by-case basis.
Statement KQ 2H: SBRT may be considered as a curative treatment option for patients with synchronous MPLC. SBRT for synchronous MPLC has equivalent rates of local control and toxicity, but decreased rates of overall survival compared with those with single tumors.
Statement KQ 2I: SBRT is recommended as a curative treatment option for patients with metachronous MPLC. SBRT for metachronous MPLC has equivalent rates of local control and toxicity and overall survival compared with those with single tumors.
For patients who underwent pneumonectomy and now have a new primary tumor in their remaining lung
Statement KQ 2J: SBRT may be considered a curative treatment option for patients with metachronous MPLC in a postpneumonectomy setting. While SBRT for metachronous MPLC appears to have equivalent rates of local control and acceptable toxicity compared to single tumors, SBRT in the post-pneumonectomy setting might have a higher rate of toxicity than in patients with higher baseline lung capacity.
KQ 3: For medically inoperable early-stage lung cancer patients, how can SBRT techniques be individually tailored to provide an adequate dose for tumor eradication with minimal risk to normal structures in “high-risk” clinical scenarios?
For tumors with intimal proximity/involvement of mediastinal structures (bronchial tree, esophagus, heart, etc.)
Statement KQ 3A: For tumors in close proximity to the proximal bronchial tree, SBRT should be delivered in 4 to 5 fractions. Physicians should endeavor to meet the constraints that have been utilized in prospective studies given the severe toxicities that have been reported.
Statement KQ 3B: For tumors in close proximity to the esophagus, physicians should endeavor to meet the constraints that have been utilized in prospective studies or otherwise reported in the literature given the severe esophageal toxicities that have been reported.
Statement KQ 3C: For tumors in close proximity to the heart and pericardium, SBRT should be delivered in 4 to 5 fractions with low incidence of serious toxicities to the heart, pericardium, and large vessels observed. Adherence to volumetric and maximum dose constraints utilized in prospective trials or reported in the literature may optimize the safety profile of this treatment.
For tumors abutting or invading the chest wall
Statement KQ 3D: SBRT is an appropriate option for treatment and should be offered for T1-2 tumors that abut the chest wall. Grade 1 and 2 chest wall toxicity is a common occurrence post SBRT that usually resolves with conservative management. Patients with peripheral tumors approximating the chest wall should be counseled on the possibility of this common toxicity.
Statement KQ 3E: SBRT may be utilized in patients with cT3 disease due to chest wall invasion without clear evidence of reduced efficacy or increased toxicity compared to tumors abutting the chest wall.
KQ 4: In medically inoperable patients, what is the role of SBRT as salvage therapy for early-stage lung cancer that recurs?
After conventionally fractionated Radiation Therapy
Statement KQ 4A: The use of salvage SBRT after primary conventionally fractionated radiation may be offered to selected patients due to reported favorable local control and survival. These patients should be informed of significant (including fatal) toxicities.
Statement KQ 4B: Patient selection for salvage SBRT after primary conventionally fractionated radiation is a highly individualized process. Radiation oncologists should assess evidence-based patient, tumor, and treatment factors prior to treatment initiation.
After SBRT and sublobar resection
Statement KQ 4C: Patient selection for salvage SBRT after previous SBRT and after prior Sublobar resection is a highly individualized process. Radiation oncologists should assess evidence-based patient, tumor, and treatment factors before treatment initiation.
Stereotactic Body Radiation Therapy for early-stage Non-Small Cell Lung Cancer: Executive Summary of an ASTRO Evidence-Based Guideline. Videtic GM, Donington J, Giuliani M, et al. http://dx.doi.org/10.1016/j.prro.2017.04.014