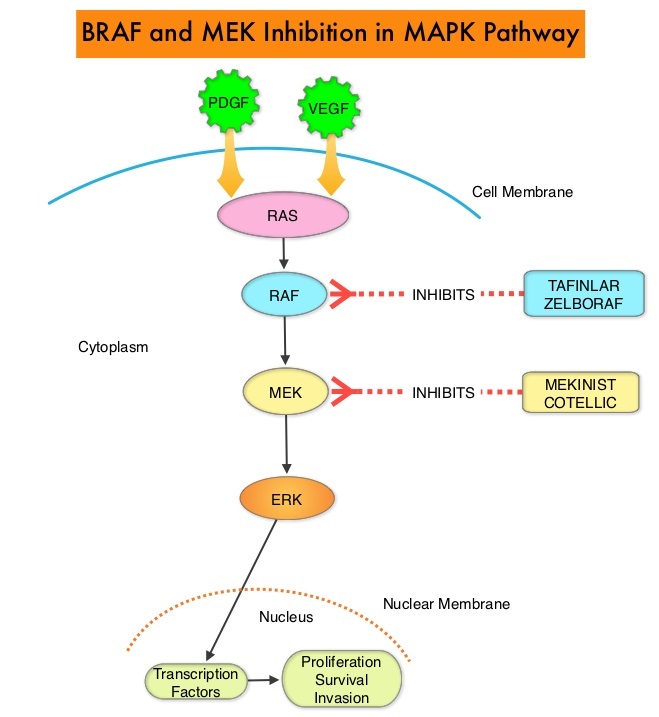
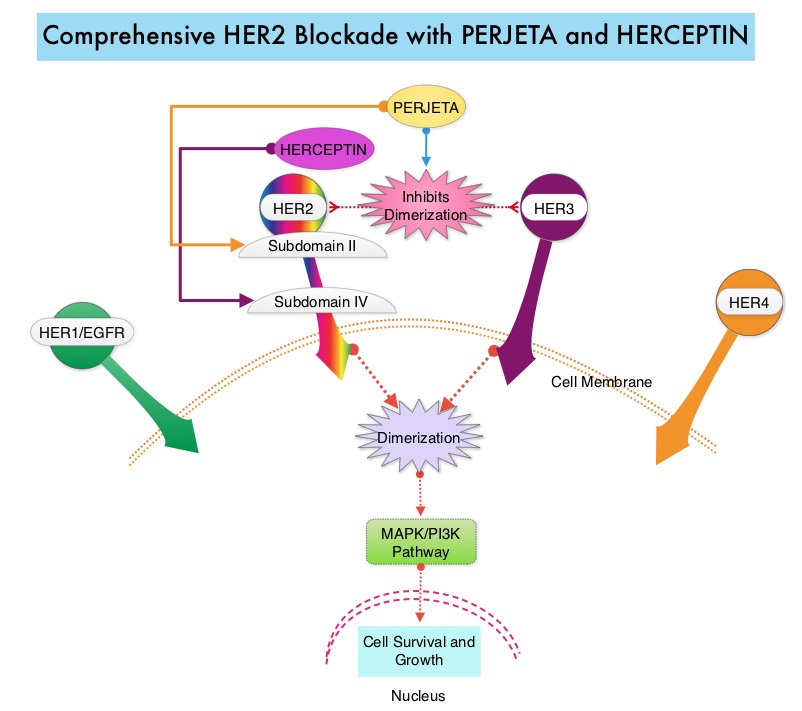
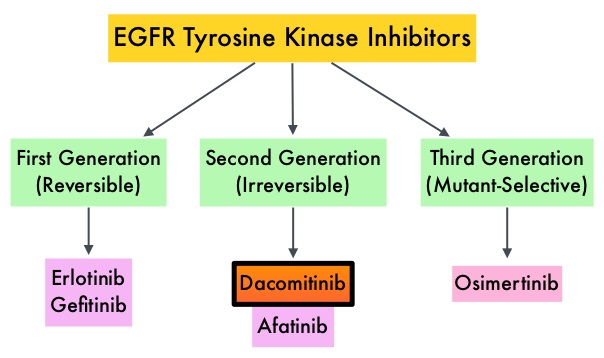
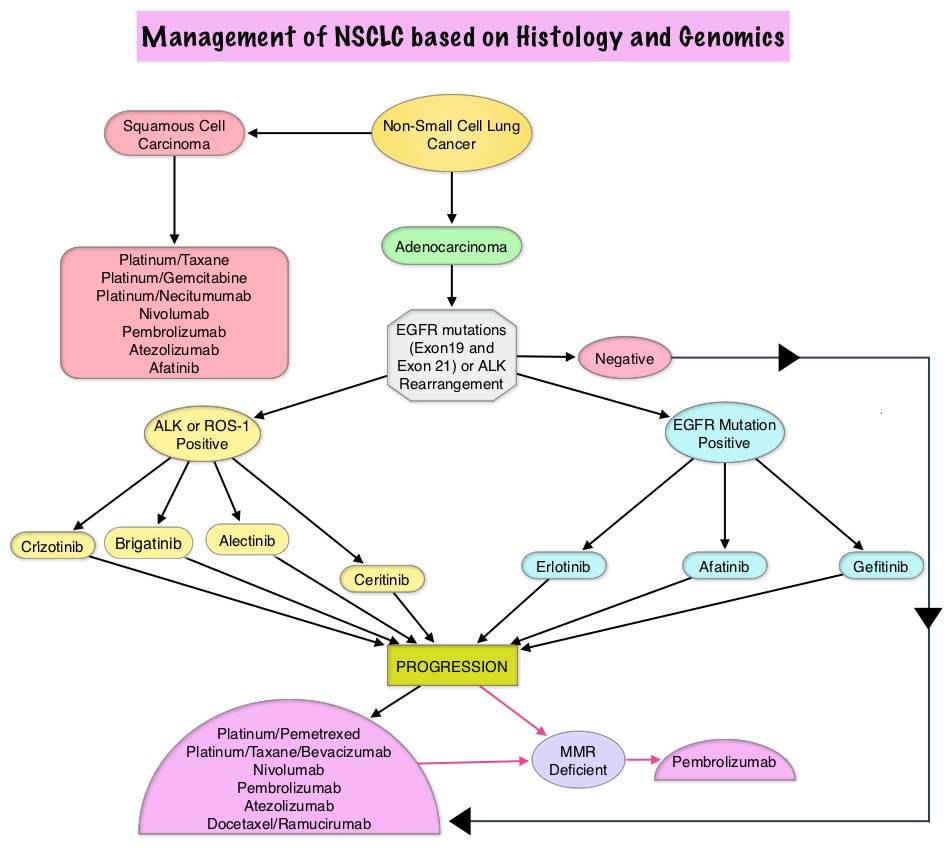
SUMMARY: The FDA on March 23, 2017, granted accelerated approval to BAVENCIO® (Avelumab) for the treatment of patients 12 years and older with metastatic Merkel Cell Carcinoma (MCC). It is estimated that about 1500 cases of MCC are diagnosed in the United States each year and the life expectancy for metastatic Merkel Cell Carcinoma is less than 1 year and is associated mortality three times that of Malignant Melanoma (46% vs. 15% respectively). Merkel Cell Carcinoma, also described as Trabecular tumor of the skin, is rare but aggressive form neuroendocrine skin cancer and is much more common in elderly Caucasians. The rapid rise in the incidence of MCC over the past several years has been attributed to increased life expectancy, more sun exposure and weakened immune systems. Approximately 80% of Merkel Cell Carcinoma tumors have been found to be infected with Merkel Cell PolyomaVirus (MCPyV) and the natural history of the MCC has been linked to virus-specific humoral and cellular immune responses. MCC tumors are able to evade the immune system in spite of persistent expression of immunogenic viral proteins. It has been postulated that a high mutation burden associated with Merkel Cell Carcinomas leads to many new antigens being presented to the immune system. Tumor cells as well as tumor-infiltrating immune cells express PD-L1 (Programmed cell Death Ligands), which can contribute to inhibition of antitumor immune response in the tumor microenvironment. The immune system is harnessed and Cytotoxic T-cell activity is suppressed by the binding of PD-L1 to PD-1(Programmed cell Death 1) and B7.1 receptors found on T cells. Merkel Cell Carcinoma is associated with increased PD-L1 expression.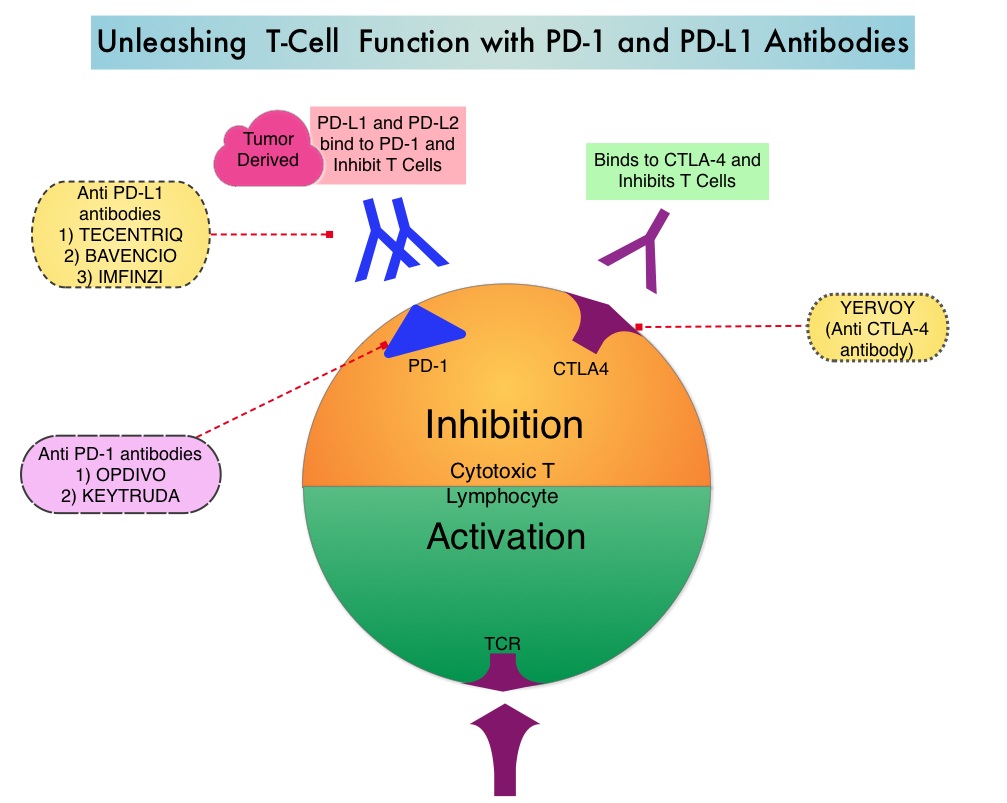
BAVENCIO® is a human, immunoglobulin G1 lambda, PD-L1 targeted monoclonal antibody that binds to PD-L1 and blocks the interaction between PD-L1 and its receptors PD-1. This in turn negates the inhibitory effects of PD-L1 on the immune response by unleashing the immune system and restoring antitumor immune responses. In addition, BAVENCIO® induces Antibody Dependent Cell-mediated Cytotoxicity (ADCC). The approval of BAVENCIO® for Merkel Cell Carcinoma was based on the phase II, prospective, open-label, international JAVELIN trial in which 88 patients with Stage IV Merkel Cell Carcinoma received BAVENCIO® 10 mg/kg IV infusion over 60 minutes, every 2 weeks, until disease progression or unacceptable toxicity. Enrolled patients had at least one prior chemotherapy regimen for metastatic disease. Over 50% of the patients had visceral metastases, two thirds of the patients had tumors with PD-L1 expression of 1% or more, by ImmunoHistoChemistry assay and 52% of the evaluable patients tested positive for Merkel cell Polyomavirus. However, patient selection in this study was not based on the level of PD-L1 expression or Polyomavirus status. The median age was 73 years. The Primary endpoint was Objective Response Rate (ORR). Secondary endpoints included Duration of Response and Progression Free Survival (PFS).
At a median follow up of 16 months, the Objective Response Rate at 1 year was 33% with a Complete Response Rate of 11%. The median time to response was 6 weeks. The 6-month durable response rate was 30.6% and the median Duration of Response had not yet been reached. These responses were noted irrespective of PD-L1 tumor cell expression or presence of Merkel cell Polyomavirus. The estimated one year PFS was 30% and one year Overall Survival was 52%. The most common adverse reactions were rash, fatigue, nausea, diarrhea, decreased appetite, musculoskeletal pain, infusion-related reaction and peripheral edema.
The authors concluded that BAVENCIO® showed durable antitumor activity with a manageable safety profile, in patients with metastatic Merkel Cell Carcinoma who had progressed on chemotherapy and is an important new treatment option for this patient population. BAVENCIO® is the very first drug approved by the FDA for Merkel Cell Carcinoma. Studies are also underway with KEYTRUDA® (Pembrolizumab), a PD-1 inhibitor, in this patient group, with promising outcomes thus far. Durable responses to avelumab (anti-PD-L1) in patients with Merkel cell carcinoma progressed after chemotherapy: 1-year efficacy update. Kaufman HL, Russell JS, Hamid O, et al. 2017 AACR Annual Meeting. Abstract CT079. Presented April 3, 2017.
 Hyaluronan is a polysaccharide present in the extracellular matrix of the subcutaneous tissue and is depolymerized by the naturally occurring enzyme Hyaluronidase.
Hyaluronan is a polysaccharide present in the extracellular matrix of the subcutaneous tissue and is depolymerized by the naturally occurring enzyme Hyaluronidase.