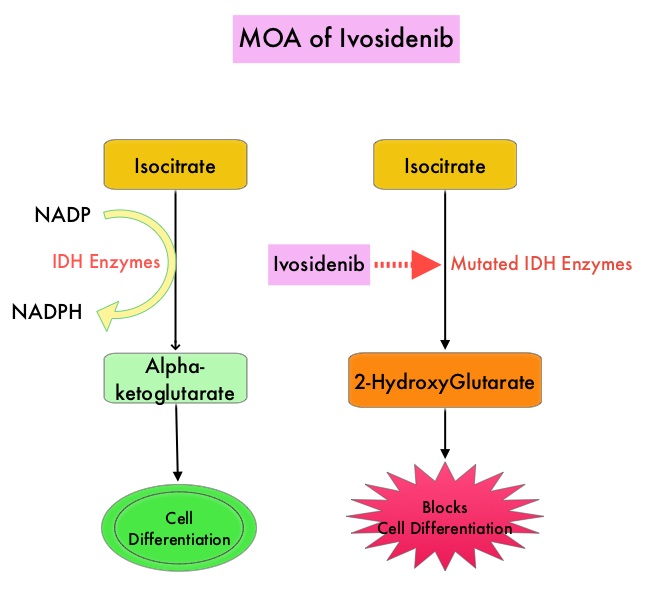
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 268,600 new cases of invasive breast cancer will be diagnosed in 2019 and about 41,760 women will die of the disease. Cyclin Dependent Kinases (CDK) play a very important role to facilitate orderly and controlled progression of the cell cycle. Genetic alterations in these kinases and their regulatory proteins have been implicated in various malignancies. Cyclin Dependent Kinases 4 and 6 (CDK4 and CDK6), phosphorylate RetinoBlastoma protein (RB), and initiate transition from the G1 phase to the S phase of the cell cycle. RetinoBlastoma protein has antiproliferative and tumor-suppressor activity and phosphorylation of RB protein nullifies its beneficial activities. CDK4 and CDK6 are activated in hormone receptor positive breast cancer, promoting breast cancer cell proliferation. Further, there is evidence to suggest that endocrine resistant breast cancer cell lines depend on CDK4 for cell proliferation. The understanding of the role of Cyclin Dependent Kinases in the cell cycle, has paved the way for the development of CDK inhibitors.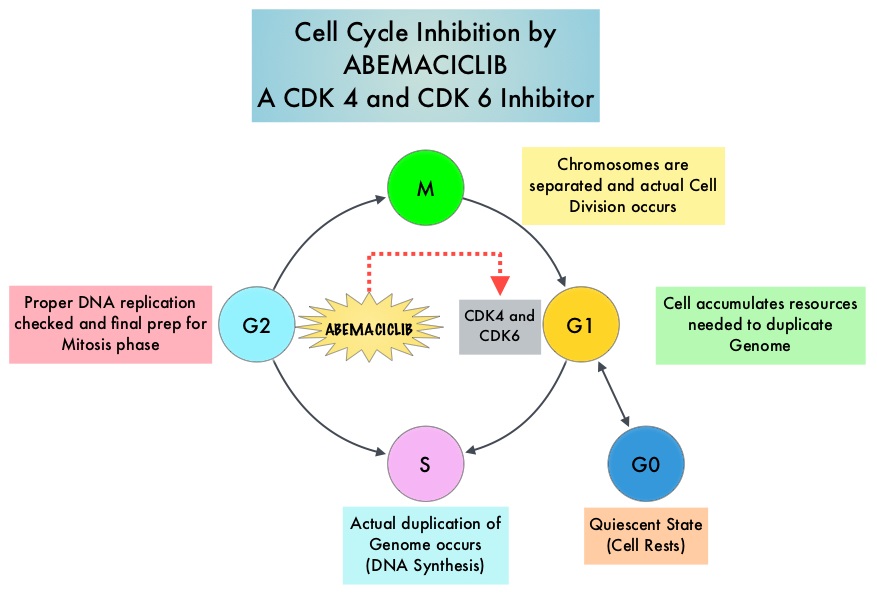
VERZENIO® (Abemaciclib) is an oral, selective inhibitor of CDK4 and CDK6 kinase activity, and prevents the phosphorylation and subsequent inactivation of the Rb tumor suppressor protein, thereby inducing G1 cell cycle arrest and inhibition of cell proliferation. VERZENIO® is structurally distinct from other CDK 4 and 6 inhibitors (such as Ribociclib and Palbociclib) and is 14 times more potent against cyclin D1/CDK 4 and cyclin D3/CDK 6, in enzymatic assays, but potentially less toxic than earlier pan-CDK inhibitors. At higher doses, only VERZENIO® causes significant cancer cell death, compared with other CDK4/6 inhibitors, suggesting that this drug may be affecting proteins, other than CDK4/6. Additionally, preclinical studies have demonstrated that VERZENIO® may have additional therapeutic benefits for a subset of tumors that are unresponsive to treatment or have grown resistant to other CDK4/6 inhibitors. It has also been shown to cross the blood-brain barrier.
The FDA approved VERZENIO® in combination with FASLODEX® (Fulvestrant) in 2017, for women with Hormone Receptor positive (HR-positive), HER2-negative, advanced or metastatic breast cancer, with disease progression following endocrine therapy. This approval was based on the MONARCH-2 study. VERZENIO® was also approved as monotherapy for women and men with HR-positive, HER2-negative advanced or metastatic breast cancer, with disease progression following endocrine therapy and prior chemotherapy in the metastatic setting, based on improved Objective Response Rate (ORR) and PFS (MONARCH-1 study).
MONARCH-2 is an International, double-blind, Phase III study in which 669 patients were randomized in a 2:1 ratio to receive either VERZENIO® plus FASLODEX® (N=446) or placebo plus FASLODEX® (N=223). Enrolled patients had HR-positive, HER2-negative metastatic breast cancer, with disease progression while receiving neoadjuvant or adjuvant endocrine therapy, within 12 months of adjuvant endocrine therapy, or while receiving endocrine therapy for metastatic breast cancer. Patients must not have received more than one endocrine therapy or any prior chemotherapy for metastatic breast cancer. Randomized patients received either VERZENIO® 150 mg or placebo orally twice daily plus FASLODEX® 500 mg IM on Day 1 and Day 15 of cycle 1 and then on Day 1 of cycle 2 and beyond (28-day cycles). Treatment was continued until disease progression or unmanageable toxicities. The mean patient age was 60 years, 82% of patients were postmenopausal, 72% had measurable disease, 56% had visceral disease, and 25% had primary endocrine therapy resistance. About 60% of patients had received chemotherapy in the adjuvant or neoadjuvant setting and 69% of the patients had prior therapy with Aromatase Inhibitors (AI). The Primary end point was Progression Free Survival (PFS), and Secondary end points included Overall Survival (OS), Objective Response Rate (ORR), Duration of Response, Clinical Benefit Rate, Quality of Life, and safety. The authors had previously reported the PFS data and the median PFS for the group receiving VERZENIO® plus FASLODEX® was 16.4 months compared with 9.3 months for those taking placebo with FASLODEX® (HR= 0.55; P<0.0001).
The authors now reported the Overall Survival (OS) data. At a median follow up of 47.7 months, the median OS with the combination of VERZENIO® plus FASLODEX® was 46.7 months compared with 37.3 months for placebo plus FASLODEX® (HR=0.757; P=0.0137). The OS benefit was consistently seen across subgroups, including those with poor prognostic factors such as visceral metastasis (HR=0.675) and primary endocrine therapy resistance (HR=0.686). The updated median PFS with VERZENIO® plus FASLODEX® was 16.9 months compared to 9.3 months with placebo plus FASLODEX® (HR=0.53; P<0.001). The time to second disease progression (PFS2) was also improved with the addition of VERZENIO® at a median 23.1 months versus 20.6 months with placebo plus FASLODEX® (HR, 0.675). VERZENIO® combination also showed a statistically significant improvement in median chemotherapy-free survival compared with placebo/FASLODEX® combination at 25.5 months and 18.2 months, respectively (HR, 0.638). The most common adverse events in the VERZENIO® versus placebo groups were diarrhea neutropenia, nausea and fatigue.
It was concluded that the addition of VERZENIO® to FASLODEX® resulted in a statistically significant improvement in Overall Survival among patients with HR-positive, HER2-negative breast cancer, who had progressed on prior endocrine therapy, significantly delaying the receipt of subsequent chemotherapy. MONARCH 2: Overall survival of abemaciclib plus fulvestrant in patients with HR+, HER2 advanced breast cancer. Sledge Jr GW, Toi M, Neven P, et al. Presented at 2019 ESMO Congress; September 27 to October 1, Barcelona, Spain. Abstract LBA6_PR.
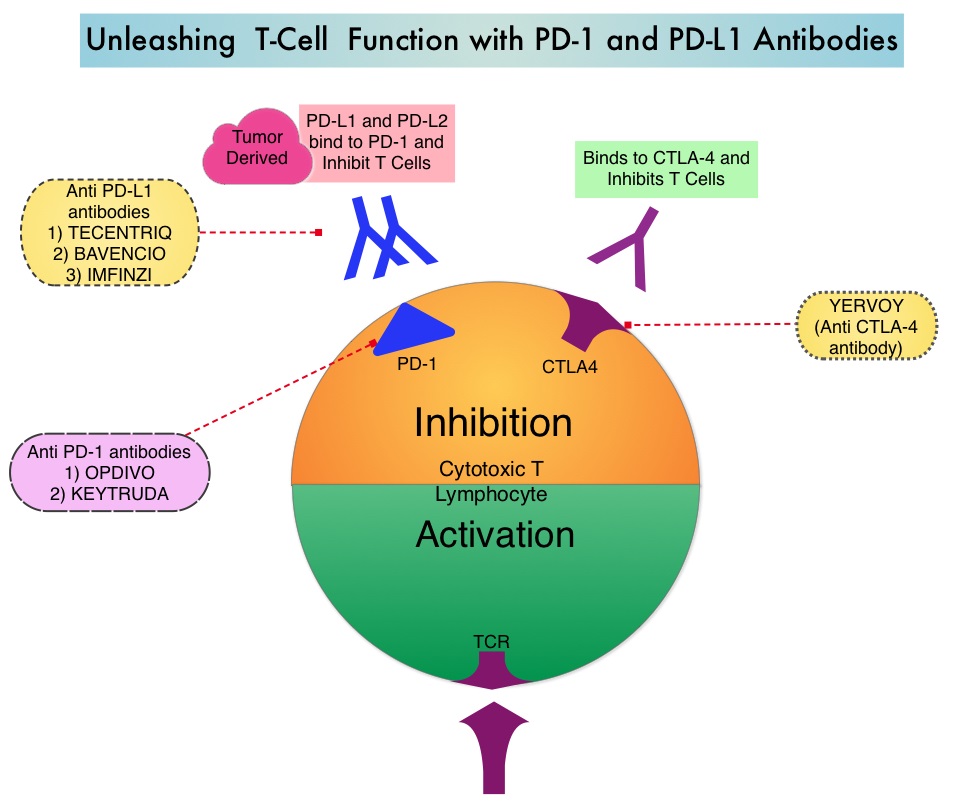
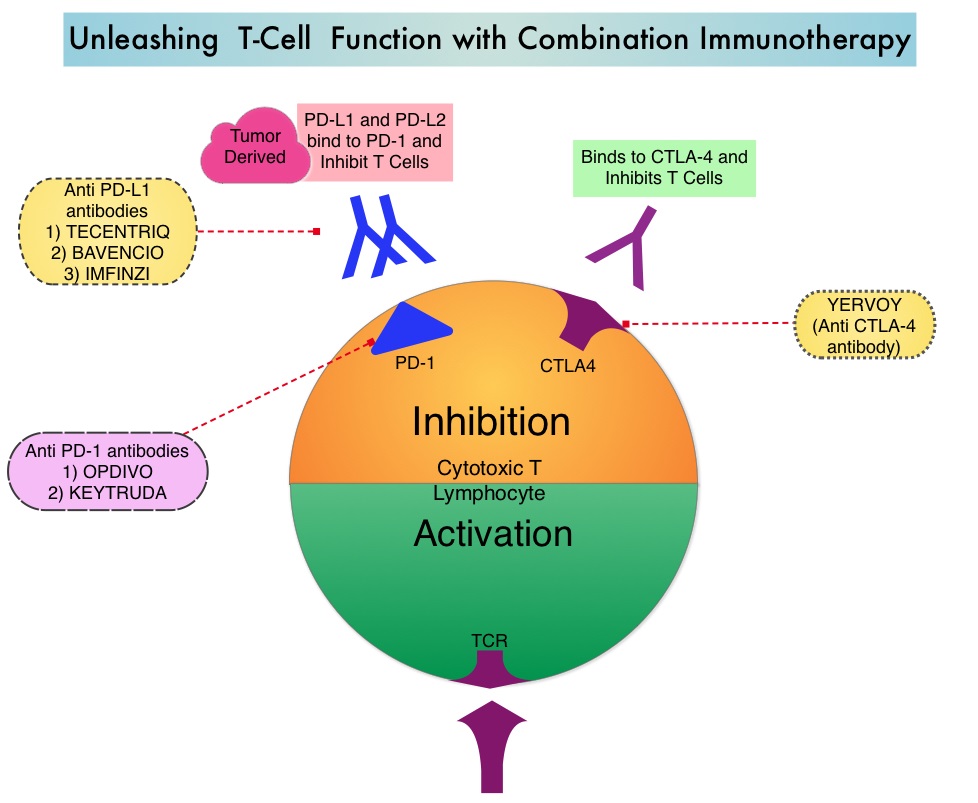
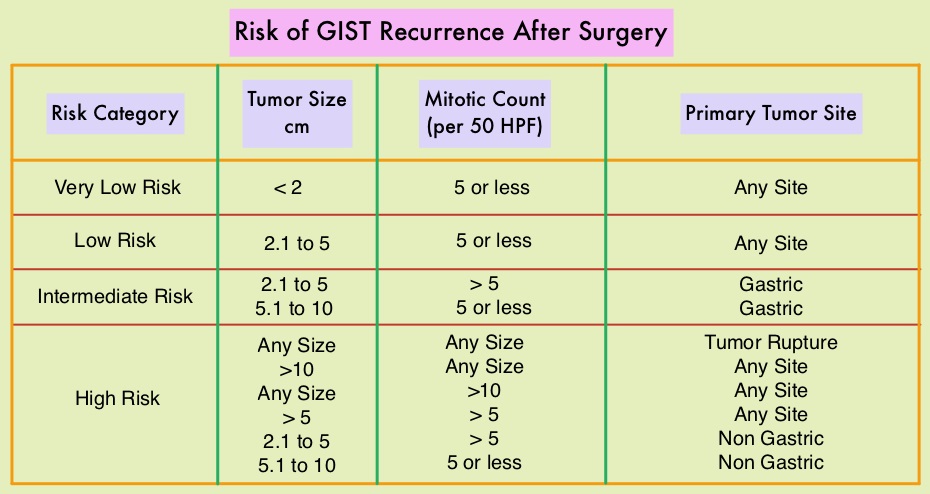 Treatment of patients with advanced or metastatic GIST with Tyrosine Kinase Inhibitor GLEEVEC® (Imatinib) achieves high Objective Response and diseases stabilization rates. Patients with KIT exon 9 mutation have a poor prognosis compared to those with KIT exon 11 mutation (most common) and benefit from a higher dose of GLEEVEC® (800 mg daily).
Treatment of patients with advanced or metastatic GIST with Tyrosine Kinase Inhibitor GLEEVEC® (Imatinib) achieves high Objective Response and diseases stabilization rates. Patients with KIT exon 9 mutation have a poor prognosis compared to those with KIT exon 11 mutation (most common) and benefit from a higher dose of GLEEVEC® (800 mg daily).