SUMMARY: The American Cancer Society estimates that in 2023, 20,380 new cases of Acute Myeloid Leukemia (AML) will be diagnosed in the United States and 11,310 patients will die of the disease. AML is one of the most common types of leukemia in adults and can be considered as a group of molecularly heterogeneous diseases with different clinical behavior and outcomes. A significant percentage of patients with newly diagnosed AML are not candidates for intensive chemotherapy or have disease that is refractory to standard chemotherapy. Even with the best available therapies, the 5-year Overall Survival in patients 65 years of age or older is less than 5%. Cytogenetic analysis has been part of routine evaluation when caring for patients with AML. By predicting resistance to therapy, tumor cytogenetics will stratify patients based on risk, and help manage them accordingly. Even though cytotoxic chemotherapy may lead to long term remission and cure in a minority of patients with favorable cytogenetics, patients with high-risk features such as unfavorable cytogenetics, molecular abnormalities, prior myelodysplasia, and advanced age, have poor outcomes with conventional chemotherapy alone. More importantly, with the understanding of molecular pathology of AML, personalized and targeted therapies are becoming an important part of the AML treatment armamentarium.
The pro-survival (anti-apoptotic) protein BCL2 is over expressed by AML cells and regulates clonal selection and cell survival. A new class of anticancer agents known as BH3-mimetic drugs mimic the activity of the physiologic antagonists of BCL2 and related proteins and promote apoptosis (programmed cell death). VENCLEXTA® (Venetoclax) is a second generation, oral, selective, small molecule inhibitor of BCL2 and restores the apoptotic processes in tumor cells. VIDAZA® (Azacitidine) is a hypomethylating agent that promotes DNA hypomethylation by inhibiting DNA methyltransferases. VIDAZA® has been shown to significantly improve Overall Survival (OS), when compared to conventional care regimens, in elderly unfit patients with newly diagnosed AML, who are not candidates for intensive chemotherapy. The combination of VIDAZA® and VENCLEXTA® in a previously published Phase Ib study was highly efficacious, with significant responses, duration of response and Overall Survival benefit.
VIALE-A is a Phase III, multicenter, randomized, double-blind, placebo-controlled confirmatory trial, conducted to evaluate the efficacy and safety of a combination of VIDAZA® and VENCLEXTA®, as compared with VIDAZA® plus placebo (the control regimen), in previously untreated patients with AML, who were ineligible for intensive induction therapy. In this study, 431 patients (N=431) with previously untreated AML were randomly assigned in a 2:1 ratio to receive either VIDAZA® plus VENCLEXTA® (N=286), or VIDAZA® plus placebo (N=145). Enrolled patients were ineligible for standard induction chemotherapy because of coexisting conditions, 75 years of age or older, or both. All patients received VIDAZA® 75 mg/m2 subcutaneously or IV on days 1 through 7 of every 28-day cycle. Patients in the study group also received VENCLEXTA® 100 mg orally on day 1 and 200 mg on day 2 and target dose of 400 mg on day 3, and continued daily until day 28 during cycle 1, to mitigate Tumor Lysis Syndrome. The dose of VENCLEXTA® was initiated at 400 mg daily in all subsequent 28-day cycles. In the control group, a matching placebo was administered orally, once daily, in 28-day cycles. The median age was 76 years in both groups, approximately 60% were male and 76% were Caucasian. Molecular abnormalities of interest included FLT-3, observed in 14% of patients receiving VIDAZA® plus VENCLEXTA®, IDH1/2, observed in 25% of patients, TP53, observed in 23.3% of patients and NPM1, observed in 16.6% of patients. Secondary AML was reported in 25% of the patients in the VIDAZA® plus VENCLEXTA® group and in 24% of the patients in the control group. All the patients were hospitalized on or before day 1 of cycle 1 and for at least 24 hours after receiving the final dose of VENCLEXTA®, in order to receive prophylaxis against the Tumor Lysis Syndrome and for monitoring. The Primary endpoint was Overall Survival (OS). The Secondary end points included Complete Remission (CR) rates, composite Complete Remission (Complete Remission or Complete Remission with incomplete hematologic recovery), RBC and platelet transfusion independence, and Quality of Life according to Patient-Reported Outcomes.
At a median follow up of 20.5 months, the median OS was 14.7 months in the VIDAZA® plus VENCLEXTA® group versus 9.6 months in the VIDAZA® plus placebo group (HR=0.66; P<0.001). VIDAZA® plus VENCLEXTA® combination resulted in a CR rate of 36.7% versus 17.9%; P<0.001 and composite CR of 66.4% versus 28.3%; P<0.001, when compared to the control regimen. Most responses were seen after the first 28-day cycle. The median time to first response was 1.3 versus and 2.8 months respectively, duration of CR was 17.5 months versus 13.3 months and median duration of composite CR was 17.5 months in the VIDAZA® plus VENCLEXTA® group and 13.4 months in the control group. RBC transfusion independence occurred in 59.8% of the patients in the VIDAZA® plus VENCLEXTA® group and in 35.2% of those in the control group (P<0.001), and platelet transfusion independence occurred in 68.5% and 49.7% (P<0.001), respectively. The benefits with VIDAZA® plus VENCLEXTA® were noted in almost all molecular subgroups compared to the control regimen. The response rates were highest among patients with FLT3 mutations (72.4% versus 36.4%, P=0.02) and those with IDH1 or IDH2 mutations (75.4 % versus 10.7%, P<0.001), respectively.
The researchers conducted 2 years of additional follow-up to determine the long-term survival benefit of VIDAZA® plus VENCLEXTA® combination and at this meeting reported the analysis of VIALE-A trial, after the occurrence of 100% of the pre-planned survival events. With a median follow-up of 43.2 months, the median Overall Survival (OS) benefit since the interim analysis in the overall population was maintained and was 14.7 months in the VIDAZA® plus VENCLEXTA® group versus 9.6 months in the VIDAZA® plus placebo group (HR=0.58; P<0.001). Among patients with Measurable Residual Disease (MRD) <10-3 who had achieved either Complete Remission (CR) or CR with incomplete hematologic recovery (CRi), the median OS was reached at 34.2 months in the VIDAZA® plus VENCLEXTA® group and 25.0 months in the control group. For patients in the IDH1/2 mutant subgroup, the median OS at final analysis with VIDAZA® plus VENCLEXTA® was 19.9 months and was 6.2 months in the control group (HR=0.31; P<0.001). Overall safety profiles were comparable between the treatment groups.
The 2-year follow up analysis of the VIALE-A trial confirmed the sustained Overall Survival benefit of VIDAZA® plus VENCLEXTA® combination in patients with AML, ineligible for intensive chemotherapy, with no new safety findings noted.
Long-Term Follow-up of the Phase 3 Viale-a Clinical Trial of Venetoclax Plus Azacitidine for Patients with Untreated Acute Myeloid Leukemia Ineligible for Intensive Chemotherapy. Pratz KW, Jonas BA, Pullarkat VA, et al. Presented at the 64th ASH Annual Meeting and Exposition, December 10-13, 2022, New Orleans, Louisiana. Abstract # 219

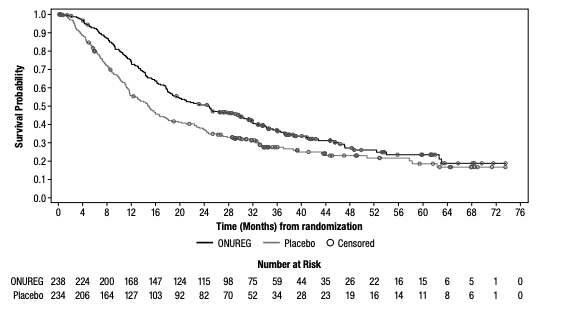
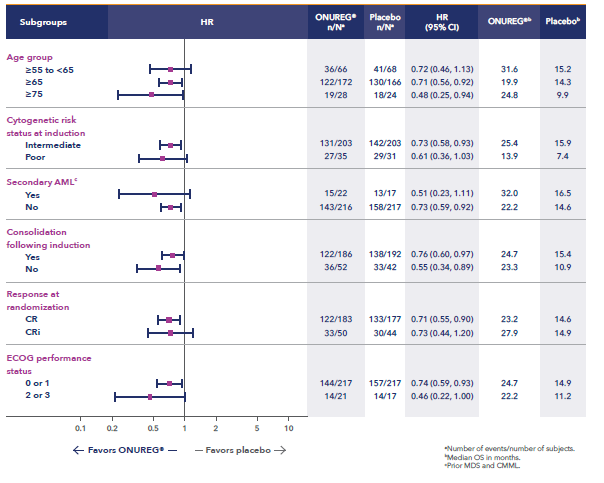
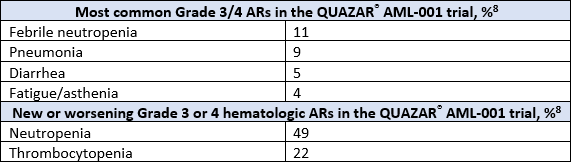
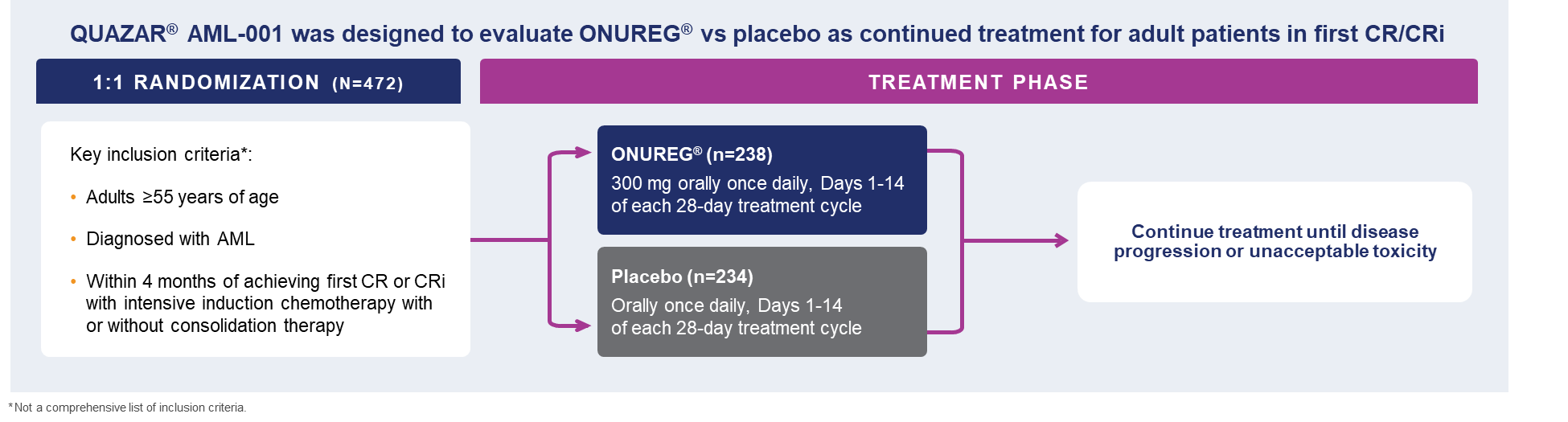 A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8
A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8