
The FDA on November 28, 2018 approved XOSPATA® for treatment of adult patients who have relapsed or refractory Acute Myeloid Leukemia (AML) with a FLT3 mutation as detected by an FDA-approved test. XOSPATA® is a product of Astellas Pharma US Inc.
Tag: Acute Myeloid Leukemia
VENCLEXTA® (Venetoclax)
The FDA on November 21, 2018 granted accelerated approval to VENCLEXTA® in combination with Azacitidine or Decitabine or low-dose Cytarabine for the treatment of newly-diagnosed Acute Myeloid Leukemia (AML) in adults who are age 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy. VENCLEXTA® is a product of AbbVie Inc. and Genentech Inc.
DAURISMO® (Glasdegib)
The FDA on November 21, 2018 approved DAURISMO® in combination with Low-Dose Cytarabine (LDAC), for newly-diagnosed Acute Myeloid Leukemia (AML) in patients who are 75 years old or older or who have comorbidities that preclude intensive induction chemotherapy. DAURISMO® is a product of Pfizer Labs.
A Completely Oral Chemotherapy-Free Regimen for Acute Promyelocytic Leukemia
SUMMARY: Acute Promyelocytic Leukemia (APL) is a subtype (M3) of Acute Myeloid Leukemia (AML) accounting for 5-10% of AMLs in adults. The diagnostic hallmark of this subtype of AML is the balanced reciprocal translocation involving the long arms of chromosomes 15 and 17 – t(15;17)(q22;q11-12), leading to the fusion of ProMyeLocytic (PML) gene with the Retinoic Acid Receptor Alpha (RARA) gene. This hybrid PML-RARA hybrid oncoprotein blocks the differentiation of promyelocytes resulting in APL. The median age for patients diagnosed with APL is around 40 years.
Patients with APL often present with life-threatening bleeding secondary to consumptive coagulopathy and more rarely thrombosis. Therefore, rapid diagnosis of APL and institution of anti-leukemic and supportive therapy is of paramount importance, to prevent bleeding related mortality. Given the early mortality rate of 17-29%, immediate institution of anti-leukemic therapy without delay is strongly recommended upon clinical suspicion of APL following morphologic evaluation of the bone marrow, pending cytogenetics. Invasive procedures, routinely done at initial presentation of AML, should be avoided. APL is a curable disease and approximately 80% of the all APL patients present with non-high risk disease (WBC 10,000 or less per microliter).
The FDA in early 2018 approved the use of TRISENOX® (Arsenic Trioxide) injection, in combination with VESANOID® (All-Trans Retinoic Acid (ATRA), Tretinoin), for the treatment of adults with newly diagnosed low-risk APL, whose APL is characterized by the presence of the t(15;17) translocation or PML/RARA gene expression. This approval was based on the superiority of Arsenic Trioxide plus ATRA which resulted in a to a 2-year Event-Free Survival (EFS) rate of 97%, compared with 86% for chemotherapy plus ATRA. The combination of chemotherapy-free ATRA and Intravenous Arsenic Trioxide is therefore considered the standard of care for non-high risk APL patients. TRISENOX® (Arsenic Trioxide) was initially approved by the FDA in 2000 for the treatment of patients with APL who are refractory or have relapsed on Retinoid and Anthracycline chemotherapy.
First published from China in the 1980’s, ATRA induces terminal differentiation of leukemic promyelocytes and leads to an immediate improvement in bleeding symptoms and almost complete resolution of the associated coagulopathy within 1-2 weeks of treatment. Arsenic Trioxide is probably the most effective single agent used in the treatment of APL. It directly binds to the PMLâ€RARA oncoprotein inducing its proteosomal degradation and leads to apoptosis of leukemic cells.
Arsenic Trioxide infusion requires hospitalization. There is however an oral tetra-arsenic tetra-sulfide (As4S4) -containing formulation, Realgar-Indigo naturalis Formula (RIF). In a previously published study, a more convenient oral RIF plus ATRA was found not to be inferior to intravenous Arsenic Trioxide plus ATRA, as first-line treatment in patients with APL (J Clin Oncol. 2013 ;31:4215-4221).
The authors in this study compared oral RIF plus ATRA treatment regimen with the standard intravenous Arsenic Trioxide plus ATRA treatment regimen in patients with non-high-risk APL. In this multicentre, non-inferiority, open-label, randomized, controlled phase III trial, patients with newly diagnosed (within 7 days) non-high-risk APL (N=109), were randomly assigned 2:1 to receive treatment with RIF-ATRA (N=72) or Arsenic Trioxide-ATRA (N=37) as the induction and consolidation therapy. Patients received RIF 60 mg/kg orally daily in divided doses or Arsenic Trioxide 0.15 mg/kg IV daily and ATRA 25 mg/m2 orally daily in divided doses. Treatment was continued until complete remission was achieved. The home-based consolidation therapy consisted of RIF 60 mg/kg orally daily in divided doses or Arsenic Trioxide 0.15 mg/kg IV daily, 4 weeks on and 4 weeks off for four cycles and ATRA 25 mg/m2 orally daily in divided doses, 2 weeks on and 2 weeks off for seven cycles. The median patient age was 35 years. The Primary outcome was Event-Free Survival at 2 years.
After a median follow up of 32 months, 97% of patients in the RIF-ATRA group and 94% in the Arsenic Trioxide-ATRA group had achieved 2-year Event-Free Survival confirming non-inferiority of RIF-ATRA compared with Arsenic Trioxide and ATRA (P=0.0017 for non-inferiority). The 2 year Overall Survival was 100% in the RIF-ATRA group and 94% in the Arsenic Trioxide-ATRA group (P=0.049). Toxicities during induction treatment included grade 3-4 hepatotoxity (elevated AST or ALT) in 9% of patients in the RIF-ATRA group versus 14% in the Arsenic Trioxide-ATRA group. Grade 3-4 infections were reported in 23% versus 42% in the two groups respectively. Two patients in the Arsenic Trioxide-ATRA group died during induction therapy (one from hemorrhage and one from thrombocytopenia).
It was concluded that oral RIF plus ATRA was not inferior to intravenous Arsenic Trioxide plus ATRA, for the treatment of patients with non-high-risk Acute Promyelocytic Leukemia. The authors suggested that this completely oral, chemotherapy-free therapy might be an alternative to the standard intravenous treatment for patients with non-high-risk APL. Oral arsenic plus retinoic acid versus intravenous arsenic plus retinoic acid for non-high-risk acute promyelocytic leukaemia: a non-inferiority, randomised phase 3 trial. Zhu HH, Wu DP, Du X, et al. Lancet Oncol. 2018;19:871-879
FDA Approves TIBSOVO® for Patients with Relapsed or Refractory Acute Myeloid Leukemia
SUMMARY: The FDA on July 20, 2018, approved TIBSOVO® (Ivosidenib) for adult patients with relapsed or refractory Acute Myeloid Leukemia (AML) with a susceptible Isocitrate DeHydrogenase-1 (IDH1) mutation, as detected by an FDA-approved test. The American Cancer Society estimates that in 2018, 19,520 new cases of Acute Myeloid Leukemia (AML) will be diagnosed in the United States and 10,670 patients will die of the disease. AML can be considered as a group of heterogeneous diseases with different clinical behavior and outcomes. Cytogenetic analysis has been part of routine evaluation when caring for patients with AML. By predicting resistance to therapy, tumor cytogenetics will stratify patients, based on risk and help manage them accordingly. Even though cytotoxic chemotherapy may lead to long term remission and cure in a minority of patients with favorable cytogenetics, patients with high risk features such as unfavorable cytogenetics, molecular abnormalities, prior myelodysplasia and advanced age, have poor outcomes with conventional chemotherapy alone.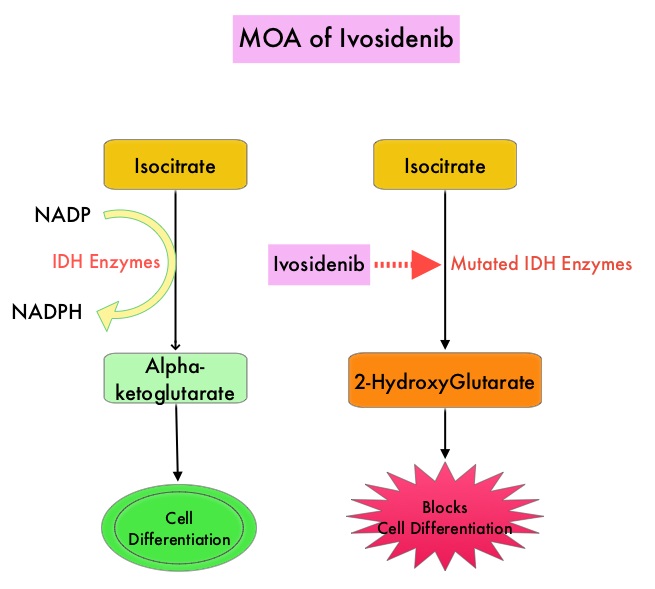
Isocitrate DeHydrogenase (IDH) is a metabolic enzyme that helps generate energy from glucose and other metabolites, by catalyzing the conversion of Isocitrate to Alpha-Ketoglutarate. Alpha-ketoglutarate is required to properly regulate DNA and histone methylation, which in turn is important for gene expression and cellular differentiation. IDH mutations lead to aberrant DNA methylation and altered gene expression thereby preventing cellular differentiation, with resulting immature undifferentiated cells. IDH mutations can thus promote leukemogenesis in Acute Myeloid Leukemia and tumorigenesis in solid tumors and can result in inferior outcomes. There are three isoforms of IDH. IDH1 is mainly found in the cytoplasm, as well as in peroxisomes, whereas IDH2 and IDH3 are found in the mitochondria, and are a part of the Krebs cycle. Approximately 20% of patients with AML, 70% of patients with Low-grade Glioma and secondary Glioblastoma, 50% of patients with Chondrosarcoma, 20% of patients with Intrahepatic cholangiocarcinoma, 30% of patients with Angioimmunoblastic T-cell lymphoma and 8% of patients with Myelodysplastic syndromes/Myeloproliferative neoplasms, are associated with IDH mutations.
TIBSOVO® is an oral, targeted, small-molecule inhibitor of mutant IDH1. IDHIFA® (Enasidenib), an oral, selective, small molecule inhibitor of mutated IDH2 protein was approved in the United States in August 2017 for adult patients with relapsed or refractory AML with an IDH2 mutation. The Complete Remission (CR) rate with the currently available non-targeted therapies for patients with relapsed or refractory AML is 15% or less and the median Overall Survival is less than 4 months, with 30-day mortality of approximately 15% and 60-day mortality of approximately 30%.
The approval of TIBSOVO® was based on an open-label, single arm, multicenter clinical trial that included 174 adult patients with relapsed or refractory AML with an IDH1 mutation. IDH1 mutations were confirmed by the Abbott RealTime® IDH1 assay, the FDA-approved test for selection of patients with AML for treatment with TIBSOVO®. TIBSOVO® was given orally at a starting dose of 500 mg daily until disease progression, unacceptable toxicity, or hematopoietic stem cell transplantation. The median treatment duration was 4.1 months. Twenty one of the 174 patients (12%) received a Stem Cell Transplant following treatment with TIBSOVO®. The median patient age was 67 years, patients had a median number of 2 prior therapies, 87% of the patients had intermediate or poor cytogenetic risk status, 33% had secondary AML and 59% were refractory to their previous therapy. The Primary endpoint was the combined rate of Complete Remission (CR) plus Complete Remission with partial hematologic recovery (CRh) and the rate of conversion from transfusion dependence to independence. CRh was defined as less than 5% of blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets more than 50,000/μl and ANC more than 500/μl)
The use of TIBSOVO® was associated with a CR plus CRh rate of 32.8%. The median time to response was 2 months and the median response duration was 8.2 months. The CR and CRh rates were 24.7% and 8.0% respectively. Among the 110 patients who were dependent on Red Blood Cell and/or platelet transfusions at baseline, 37.3% became transfusion independent during any 56-day post-baseline period. Of the 64 patients who were independent of both RBC and platelet transfusions at baseline, 59.4% remained transfusion independent during any 56-day post-baseline period. The most common adverse reactions were fatigue, nausea, diarrhea, rash, pyrexia, arthralgia, leukocytosis and QT prolongation. One effect of the IDH inhibitors are induction of differentiation of the malignant cells, and in 10-20% of patients, a clinical syndrome known as the IDH differentiation syndrome can occur. The IDH differentiation syndrome should be promptly managed by dose interruption and treatment with glucocorticoids, oral hydroxyurea, or both.
It was concluded that in patients with advanced IDH1-mutated relapsed or refractory AML, TIBSOVO® fills an unmet need, with durable molecular remissions and reduction in the need for transfusion support. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. DiNardo CD, Stein EM, de Botton S, et al. N Engl J Med 2018; 378:2386-2398
TIBSOVO® (Ivosidenib)
The FDA on July 20, 2018 approved TIBSOVO® for adult patients with relapsed or refractory Acute Myeloid Leukemia (AML) with a susceptible IDH1 mutation, as detected by an FDA-approved test. TIBSOVO® is a product of Agios Pharmaceuticals, Inc.
MYLOTARG® (Gemtuzumab ozogamicin)
The FDA on September 1, 2017 approved MYLOTARG® for the treatment of newly-diagnosed CD33-positive Acute Myeloid Leukemia (AML) in adults and for treatment of relapsed or refractory CD33-positive AML in adults and in pediatric patients 2 years and older. MYLOTARG® may be used in combination with Daunorubicin and Cytarabine for adults with newly diagnosed AML, or as a stand-alone treatment for certain adult and pediatric patients. MYLOTARG® is a product of Pfizer Inc.
FDA Approves IDHIFA® for Patients with Relapsed or Refractory Acute Myeloid Leukemia
SUMMARY: The FDA on August 1, 2017 granted regular approval to IDHIFA® (Enasidenib), for the treatment of adult patients with relapsed or refractory Acute Myeloid Leukemia (AML) with an Isocitrate DeHydrogenase-2 (IDH2) mutation, as detected by an FDA-approved test. The American Cancer Society estimates that in 2017, 21,380 new cases of Acute Myeloid Leukemia (AML) will be diagnosed in the United States and 10,590 patients will die of the disease. AML can be considered as a group of heterogeneous diseases with different clinical behavior and outcomes. Cytogenetic analysis has been part of routine evaluation when caring for patients with AML. By predicting resistance to therapy, tumor cytogenetics will stratify patients, based on risk and help manage them accordingly. Even though cytotoxic chemotherapy may lead to long term remission and cure in a minority of patients with favorable cytogenetics, patients with high risk features such as unfavorable cytogenetics, molecular abnormalities, prior myelodysplasia and advanced age, have poor outcomes with conventional chemotherapy alone.
Isocitrate DeHydrogenase (IDH) is a metabolic enzyme that helps generate energy from glucose and other metabolites by catalyzing the conversion of Isocitrate to Alpha-Ketoglutarate. Alpha-ketoglutarate is required to properly regulate DNA and histone methylation, which in turn is important for gene expression and cellular differentiation. IDH mutations lead to aberrant DNA methylation and altered gene expression thereby preventing cellular differentiation, with resulting immature undifferentiated cells. IDH mutations may thus promote leukemogenesis in Acute Myeloid Leukemia and tumorigenesis in solid tumors. There are three isoforms of IDH. IDH1 is mainly found in the cytoplasm, as well as in peroxisomes, whereas IDH2 and IDH3 are found in the mitochondria, and are a part of the Krebs cycle. Approximately 20% of patients with AML, 70% of patients with Low-grade Glioma and secondary Glioblastoma, 50% of patients with Chondrosarcoma, 20% of patients with Intrahepatic cholangiocarcinoma, 30% of patients with Angioimmunoblastic T-cell lymphoma and 8% of patients with Myelodysplastic syndromes/Myeloproliferative neoplasms, are associated with IDH mutations.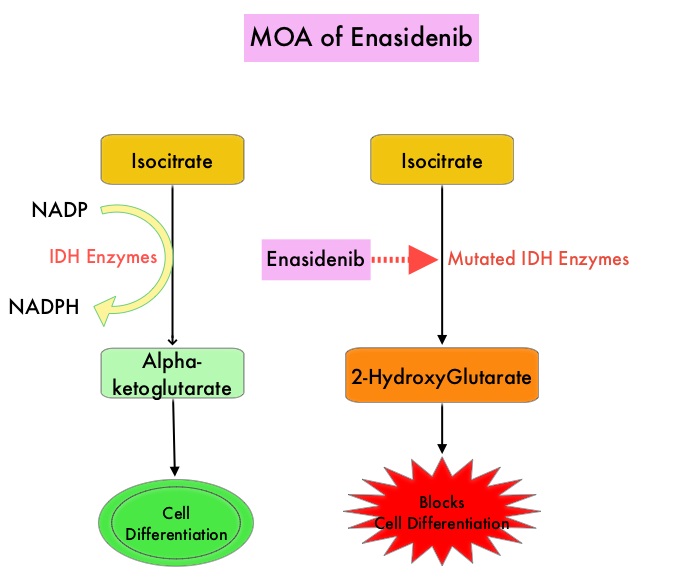
IDHIFA® is an oral, selective, small molecule inhibitor of mutated IDH2 protein. The approval of IDHIFA® was based on an open label, single arm, multicenter, clinical trial that included 199 adults with relapsed or refractory AML, who had an IDH2 mutation as detected by the RealTime IDH2 Assay. Patients received IDHIFA® 100 mg orally daily. The median age was 67 years, the median number of prior therapies was 2 and a third of the patients had unfavorable cytogenetics. Study endpoints included Complete Response (CR) and Complete Response with partial hematologic recovery (CRh) rates, CR/CRh duration, and conversion from transfusion dependence to transfusion independence.
After a median follow up of 6.6 months, 23% of patients experienced CR or CRh lasting a median of 8.2 months, with 19% of patients having a CR lasting a median 8.2 months, and 4% with a CRh lasting a median 9.6 months. The median time to first response was 1.9 months and the median time to best response of CR/CRh was 3.7 months. Of the 157 patients who required transfusions at the initiation of the trial, 34% of the patients no longer required transfusions during at least one 8 week time period on IDHIFA®. Of the 42 patients who did not require transfusions at the start of the study, 76% maintained transfusion independence. The most common toxicities were nausea, vomiting, diarrhea, elevated bilirubin and decreased appetite. Differentiation syndrome occurred in 14% of patients and these patients should be promptly managed, as this could be fatal.
The authors concluded that IDHIFA® is well tolerated and induced lasting Complete Responses in patients who had failed prior AML therapies, with the clinical efficacy related to differentiation of myeloblasts rather than cytotoxicity. This is the first FDA approval for relapsed or refractory AML specifically with an IDH2 mutation. Enasidenib in mutant-IDH2 relapsed or refractory acute myeloid leukemia (R/R AML): Results of a phase I dose-escalation and expansion study. Stein EM, Dinardo CD, Pollyea DA, et al. J Clin Oncol 35, 2017 (suppl; abstr 7004).
VYXEOS ® (Liposome-encapsulated combination of Daunorubicin and Cytarabine)
The FDA on August 3, 2017 granted regular approval to VYXEOS ® for the treatment of adults with newly-diagnosed therapy-related AML (t-AML) or AML with Myelodysplasia-Related Changes (AML-MRC), two types of AML having a poor prognosis. VYXEOS ® is a product of Jazz Pharmaceuticals.
IDHIFA® (Enasidenib)
The FDA on August 1, 2017 granted regular approval to IDHIFA®, for the treatment of adult patients with relapsed or refractory Acute Myeloid Leukemia with an Isocitrate DeHydrogenase-2 (IDH2) mutation, as detected by an FDA-approved test. IDHIFA® is a product of Celgene Corp.