SUMMARY: The DNA MisMatchRepair (MMR) system plays a crucial role in repairing DNA replication errors in normal and cancer cells. It is responsible for molecular surveillance and works as an editing tool that identifies errors within the microsatellite regions of DNA and removes them. Defective MMR system leads to MSI (Micro Satellite Instability) and accumulation of mutations (hypermutation) and the generation of neoantigens, triggering an enhanced antitumor immune response.
MSI is therefore a hallmark of defective/deficient DNA MisMatchRepair (dMMR) system. Defective MMR can be a sporadic or heritable event. Defective MMR can manifest as a germline mutation occurring in MMR genes including MLH1, MSH2, MSH6 and PMS2. This produces Lynch Syndrome often called Hereditary Nonpolyposis Colorectal Carcinoma – HNPCC, an Autosomal Dominant disorder that is often associated with a high risk for Colorectal and Endometrial carcinoma, as well as several other malignancies including Ovary, Stomach, Small bowel, Hepatobiliary tract, Brain and Skin. MSI is a hallmark of Lynch Syndrome-associated cancers. MSI tumors tend to have better outcomes and this has been attributed to the abundance of Tumor Infiltrating Lymphocytes in these tumors from increase immunogenicity. These tumors therefore are susceptible to blockade with Immune Checkpoint Iinhibitors (ICIs). The positive outcomes following ICI treatment in MSI-H tumors may be related to the possible association with Programmed Death-Ligand 1 (PD-L1) expression and the high Tumor Mutational Burden (TMB) of these diseases.
Immunotherapy with Immune Checkpoint Inhibitors (ICIs) has revolutionized cancer care and has become one of the most effective treatment options, by improving Overall Response Rate and prolongation of survival, across multiple tumor types. These agents target Programmed cell Death protein-1 (PD-1), Programmed cell Death Ligand-1 (PD-L1), Cytotoxic T-Lymphocyte-Associated protein-4 (CTLA-4), and many other important regulators of the immune system. Checkpoint inhibitors unleash the T cells resulting in T cell proliferation, activation, and a therapeutic response.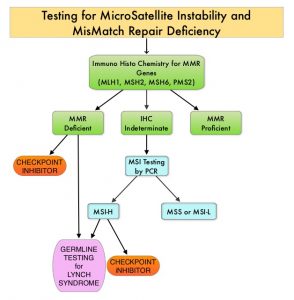
MSI testing is performed using a PCR based assay and MSI-High refers to instability at 2 or more of the 5 mononucleotide repeat markers and MSI-Low refers to instability at 1 of the 5 markers. Patients are considered Micro Satellite Stable (MSS) if no instability occurs. MSI-L and MSS are grouped together because MSI-L tumors are uncommon and behave similar to MSS tumors. Tumors considered MSI-H have deficiency of one or more of the DNA MMR genes. MMR gene deficiency can be detected by ImmunoHistoChemistry (IHC).
The authors in this publication conducted a systematic review and meta-analysis which included a total of 14 published articles that evaluated ICIs in the treatment of advanced MSI-H tumors from inception to December 2019. These articles were identified by searching the PubMed, EMBASE, and Cochrane Library databases. Overall, 939 patients in the 14 studies were analyzed, and the purpose of this study was to determine the outcomes in patients with advanced, MSI-H cancers, following treatment with ICIs. The selected studies for analysis had prospectively accrued patients with advanced or metastatic MSI-H/dMMR cancers, regardless of line of therapy, and data was available for Overall Response Rate (ORR) and/or survival analysis (Overall Survival and/or Progression Free Survival).
The studies included use of either, Avelumab (BAVENCIO®), Pembrolizumab (KEYTRUDA®), Ipilimumab (YERVOY®), Nivolumab (OPDIVO®), Atezolizumab (TECENTRIQ®) or Durvalumab (IMFINZI®). This analysis included a range of tumor types, and the Primary outcome of interest was Overall Response Rate (ORR). Secondary end points were median Progression Free Survival (PFS), median Overall Survival (OS), pooled rate of patients alive at 1, 2 and 3 years, and pooled rate of patients that attained Disease Control Rate (DCR), which is the sum of Stable Disease rate and ORR.
The pooled ORR was 41.5%, the pooled DCR was 62.8%, the pooled median PFS was 4.3 months and the pooled median OS was 24 months. The pooled 1 and 2-year OS were 75.6% and 56.5% respectively. Since only one study provided 3-year OS data, a formal pooled analysis for 3 years was not possible. The ORR was similar according to histologic analysis with the higher values for Gastric cancer (61.2%) and the lowest ORR associated with Colorectal cancer (47.1%), Endometrial (36.1%), and other tumors (35.5%).
It was concluded from this meta-analysis that Immune Checkpoint Inhibitors were associated with high activity, independent of tumor type and drug used, and molecular biomarkers such as MisMatch Repair proteins may have a predictive value for the activity of immunotherapy.
Outcomes Following Immune Checkpoint Inhibitor Treatment of Patients With Microsatellite Instability-High Cancers. A Systematic Review and Meta-analysis. Petrelli F, Ghidini M, Ghidini A, et al. JAMA Oncol. 2020;6:1068-1071.