
The FDA on September 26, 2019 approved DARZALEX® for adult patients with Multiple Myeloma in combination with VELCADE® (Bortezomib), THALOMID® (Thalidomide), and Dexamethasone in newly diagnosed patients who are eligible for Autologous Stem Cell Transplant (ASCT). DARZALEX® is a product of Janssen Biotech, Inc.
Tag: Multiple Myeloma
XPOVIO® (Selinexor)
The FDA on July 3, 2019 granted accelerated approval to XPOVIO® in combination with Dexamethasone for adult patients with Relapsed or Refractory Multiple Myeloma (RRMM) who have received at least four prior therapies, and whose disease is refractory to at least two Proteasome Inhibitors, at least two Immunomodulatory agents, and an anti-CD38 monoclonal antibody. XPOVIO® is a product of Karyopharm Therapeutics.
DARZALEX® (Daratumumab)
The FDA on June 27, 2019 approved DARZALEX® in combination with REVLIMID® (Lenalidomide) and Dexamethasone for patients with newly diagnosed Multiple Myeloma who are ineligible for Autologous Stem Cell Transplant. DARZALEX® is a product of Janssen Biotech, Inc.
Anti-BCMA CAR T-Cell Therapy in Relapsed or Refractory Multiple Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 32,110 new cases will be diagnosed in 2019 and 12,960 patients are expected to die of the disease. Multiple Myeloma (MM) in 2019 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Almost all patients eventually will relapse, and patients with a high-risk cytogenetic profile or refractory disease have the worst outcomes.
Chimeric Antigen Receptor (CAR) T-cell therapy has been associated with long-term disease control in some hematologic malignancies and is emerging as a novel treatment for patients with Relapsed and Refractory Multiple Myeloma. B-cell Maturation Antigen (BCMA) is a member of the Tumor Necrosis Factor superfamily of proteins. It is a transmembrane signaling protein primarily expressed by malignant and normal plasma cells and some mature B cells. BCMA is involved in JNK and NF-kB signaling pathways that induce B-cell development and autoimmune responses. BCMA has been implicated in autoimmune disorders as well as B-lymphocyte malignancies, Leukemia, Lymphomas, and Multiple Myeloma.
Anti-BCMA CAR T-Cell Therapy bb2121 is a type of immunotherapy and consists of T cells collected from the patient’s blood in a leukapheresis procedure. These T cells are then stimulated by treating with interleukin 2 (IL-2) and anti-CD3 antibodies in vitro, so that they will actively proliferate and expand to large numbers. These T cells are then genetically engineered to produce special receptors on their surface called Chimeric Antigen Receptors (CAR), by transducing with a gene encoding the engineered CAR, via a retroviral vector such as lentiviral vector. These reprogrammed cytotoxic T cells with the Chimeric Antigen Receptors on their surface are now able to recognize a specific antigen such as BCMA on tumor cells. These genetically engineered and reprogrammed CAR T-cells are grown in the lab and are then infused into the patient. These cells in turn proliferate in the patient’s body and the engineered receptor on the cell surface help recognize and kill cancer cells that expresses that specific antigen such as BCMA. The patient undergoes lymphodepletion chemotherapy with Fludarabine and Cytoxan prior to the introduction of the engineered CAR T-cells. By depleting the number of circulating leukocytes, cytokine production is upregulated and reduces competition for resources, which in turn promotes the expansion of the engineered CAR T-cells. In a mouse model of human Multiple Myeloma, a single-dose administration of bb2121 showed rapid and durable tumor responses, with 100% survival. On the basis of these findings, the authors conducted a Phase 1 clinical study (CRB-401) of bb2121 involving patients with Relapsed or Refractory Multiple Myeloma and reported the initial results from this ongoing study.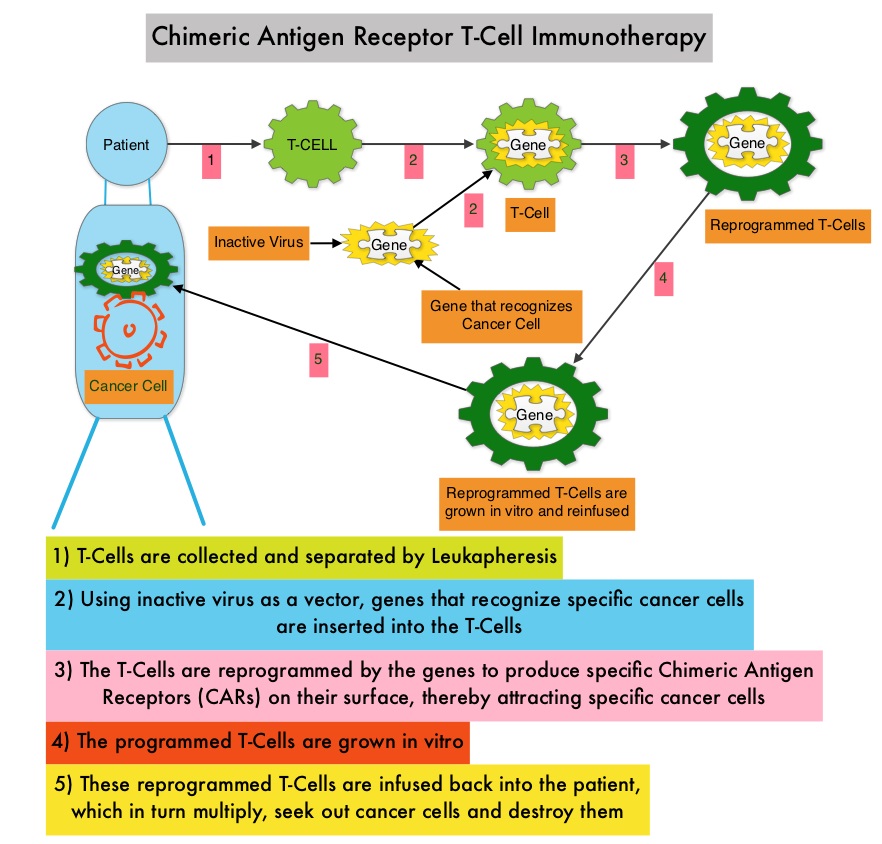
A total of 36 consecutive patients were enrolled in the study and underwent leukapheresis. Patients received bb2121 as a single infusion at increasing doses in the dose-escalation phase (50×106, 150×106, 450×106, or 800×106 CAR T-cells) followed by a dose expansion phase. The median number of previous treatment regimens was 7, and most of the enrolled patients had received previous Autologous Stem-Cell Transplantation. Further, all the patients had previously received both VELCADE® (Bortezomib) and REVLIMID® (Lenalidomide), and more than 75% of patients were exposed to VELCADE®, KYPROLIS® (Carfilzomib), REVLIMID®, POMALYST® (Pomalidomide) and DARZALEX® (Daratumumab). The median patient age was 60 years and the median time from diagnosis was 5 years. Approximately two thirds of the patients had Stage II or III disease, 27% had extramedullary disease, and 45% had a high-risk cytogenetic profile, defined by the presence of del(17p), t(4;14), or t(14;16). The Primary end point was Safety. The median duration of follow up after bb2121 infusion was 11.3 months.
Hematologic Grade 3 Adverse Events manifesting as cytopenias were the most common. Approximately 70% of patients had Grade 1 or 2 Cytokine Release Syndrome (CRS) and 6% had Grade 3 CRS. Approximately 40% of patients had Grade 1 or 2 neurotoxicity.
The Objective Response Rate was 85%, with 45% Complete Responses. The median time to first Partial Response or better was 30 days. The median Duration of Response was 10.9 months. Response rates were independent of tumor BCMA expression. All patients who had a Partial Response or better and who could be evaluated for Minimal Residual Disease (MRD) had MRD-negative status (10−4 or less nucleated cells). CAR T-cells persisted for up to 1 year after the infusion and CAR T-cell expansion was associated with responses. The median Progression Free Survival was 11.8 months.
It was concluded that, CAR T-cell therapy with bb2121 showed promising efficacy at dose levels of 150×106 or more CAR T-Cells, in a heavily pretreated population of patients with Multiple Myeloma. The authors added that non-hematologic toxicities were grade 2 or lower. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. Raje N, Berdeja J, Lin Y, et al. N Engl J Med 2019;380:1726-1737
Maintenance Therapy with NINLARO® Extends Progression Free Survival in Multiple Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 32,110 new cases will be diagnosed in 2019 and 12,960 patients are expected to die of the disease. Multiple Myeloma (MM) in 2019 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Maintenance or Continuous Treatment in patients with newly diagnosed multiple myeloma following induction and consolidation, can result in significantly longer PFS and OS, compared to those patients who receive therapy for a fixed duration of time. REVLIMID® (Lenalidomide) was approved by the FDA in 2017 as maintenance therapy for patients with multiple myeloma following Autologous Stem Cell Ttransplant (ASCT) and to date is the only drug approved for this indication. REVLIMID® maintenance however is associated with the development of second new primary malignancies and tolerability issues.
Proteasomes are enzymes found in cells and they enable the breakdown of abnormal or mutant proteins. The amino acids from these proteins are recycled to make new proteins. Just like normal cells make proteins, so do cancerous cells. But the proteins made by the cancerous cells are ineffective and in excess. Myeloma cells depend on the Proteasomes to facilitate this metabolic function, to regulate their growth and survival. Proteasome Inhibitors (PIs) inhibit Proteasome function and are a backbone of multiple myeloma treatment. VELCADE® (Bortezomib), a Proteasome Inhibitor has shown promising activity in early clinical trials, as maintenance treatment post-ASCT. The limitations with VELCADE® as maintenance therapy include, parenteral administration and tolerability. There is therefore an unmet need for an effective oral PI maintenance therapy that is convenient for the patients, with acceptable toxicities. NINLARO® (Ixazomib) unlike VELCADE® (Bortezomib) is a second generation, oral, Proteasome Inhibitor, which disrupts protein metabolism in myeloma cells, by inhibiting Proteasomes and has an antiproliferative and pro-apoptotic effect.
TOURMALINE-MM3 study is a multicenter, double-blind, placebo-controlled, phase III trial in which weekly NINLARO® was compared with placebo, as maintenance treatment, in newly diagnosed multiple myeloma patients, who had at least a Partial Response to induction therapy with a Proteasome Inhibitor and/or Immunomodulatory drug, (IMiD) followed by single Autologous Stem Cell Transplantation (ASCT). In this study, 656 patients were randomized in a 3:2 ratio to receive NINLARO® (N=395) at a dose of 3 mg orally during cycles 1-4, increasing to 4 mg from cycle 5 (if tolerated during previous cycles) or matched placebo (N=261), on days 1, 8, and 15 of 28-day cycles, for up to 2 years or until progressive disease or unacceptable toxicity. Both treatment groups were well balanced. The median age was 57 years and 37% had International Staging System (ISS) stage I disease and 63% had ISS stage II or III disease. About 18% of patients had high-risk cytogenetics such as del(17p), t(4;14), or t(14;16) and close to 90% of patients had received induction therapy with a Proteasome Inhibitor prior to ASCT. Patients were ineligible if they had received post-ASCT consolidation or tandem ASCT. The Primary endpoint was Progression Free Survival per Independent Review Committee (IRC), who were blinded to treatment assignment. The key Secondary endpoint was Overall Survival. The authors herein reported the data from the final analysis for Progression Free Survival.
After a median follow up of 31 months, the median PFS was 26.5 months with NINLARO® versus 21.3 months with placebo (HR=0.72; P=0.002). This corresponded to a 39% improvement in PFS and 28% reduction in the risk of progression or death, meeting the Primary endpoint of this study. The PFS benefit was observed broadly across patient subgroups. NINLARO® maintenance led to higher rates of deep response compared with placebo (P=0.004) and there was a higher rate of conversion from documented MRD positivity at study entry to MRD negativity with NINLARO®, compared with placebo (12% versus 7%). Overall Survival has not yet been reached in both treatment groups. Grade 3 or more Adverse Events were more common with NINLARO® (19%) versus placebo (5%), and overall 7% of patients on NINLARO® discontinued treatment compared with 5% on placebo. There was no difference in the rate of new second primary malignancies and was 3% in both arms. Further Quality of Life scores were similar in the two treatment groups.
It was concluded that NINLARO® maintenance in responding patients after ASCT resulted in a significant reduction in the risk of progression and death, and was associated with a favorable safety profile, including an absence of risk of second primary malignancies and low rates of peripheral neuropathy. The authors added that NINLARO® has a different mechanism of action and provides an alternative to REVLIMID®. With its manageable toxicity profile and convenient weekly oral dosing, NINLARO® would be ideal for maintenance treatment. Maintenance Therapy with the Oral Proteasome Inhibitor (PI) Ixazomib Significantly Prolongs Progression-Free Survival (PFS) Following Autologous Stem Cell Transplantation (ASCT) in Patients with Newly Diagnosed Multiple Myeloma (NDMM): Phase 3 Tourmaline-MM3 Trial. Dimopoulos MA, Gay F, Schjesvold FH, et al. Proceedings from the 2018 ASH Annual Meeting and Exposition; December 1 to 4, 2018; San Diego, California. Abstract 301.
Late Breaking Abstract – ASH 2018 Frontline DARZALEX® with REVLIMID® and Dexamethasone – A New Standard for Transplant-Ineligible Myeloma Patients
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma (MM) in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). REVLIMID® (Lenalidomide) based regimens are often prescribed for patients with newly diagnosed, transplant-ineligible Multiple Myeloma. REVLIMID®, a thalidomide analogue has immunomodulatory, tumoricidal, and antiangiogenic properties, and synergizes with Dexamethasone to enhance anti-myeloma activity. DARZALEX® (Daratumumab) is a human IgG1 monoclonal antibody that targets CD38, a transmembrane glycoprotein abundantly expressed on malignant plasma cells and with low levels of expression on normal lymphoid and myeloid cells. DARZALEX® exerts its cytotoxic effect on myeloma cells by multiple mechanisms, including Antibody Dependent Cellular Cytotoxicity (ADCC), Complement Mediated Cytotoxicity and direct apoptosis. Additionally, DARZALEX® may have a role in immunomodulation by depleting CD38-positive regulator Immune suppressor cells, and thereby expanding T cells, in patients responding to therapy.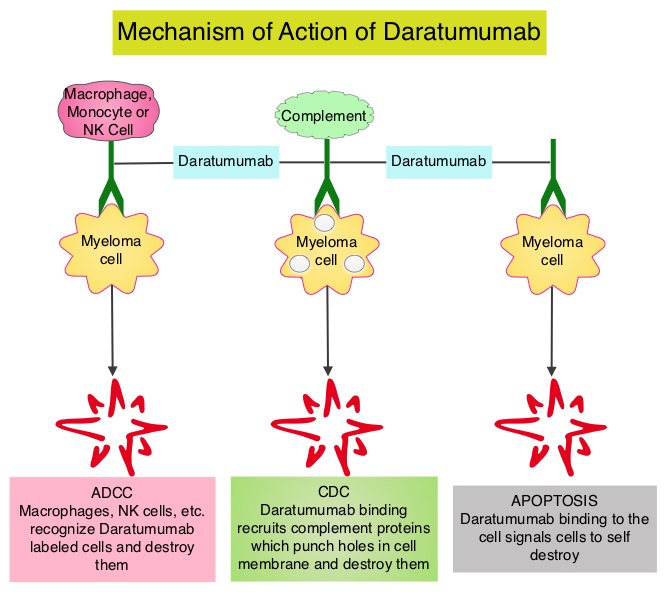
The FDA in May, 2018 approved DARZALEX® in combination with VELCADE® (Bortezomib), a proteasome inhibitor, Melphalan, an alkylating agent and Prednisone (VMP regimen), for the treatment of patients with newly diagnosed Multiple Myeloma who are ineligible for Autologous Stem Cell Transplant (ASCT). VMP regimen however is mostly utilized in Europe and not in the US. In the POLLUX trial, addition of DARZALEX® to REVLIMID® and Dexamathasone (D-Rd) showed the greatest benefit, with a 63% reduction in risk of disease progression or death (HR=0.37; P<0.001) in patients with Multiple Myeloma who had at least one prior line of therapy, compared to REVLIMID® and Dexamathasone (Rd) . Based on the efficacy and tolerable safety profile of D-Rd, the authors conducted a phase III study (MAIA), comparing D-Rd to Rd in transplant-ineligible newly diagnosed Multiple Myeloma patients and reported the prespecified interim analysis of the study.
The MAIA study is a multicenter, international, open-label, phase III trial, which included 737 newly diagnosed Myeloma patients who were not candidates for high-dose chemotherapy and Autologous Stem Cell Transplant (ASCT), due to age 65 years or older or comorbidities. Patients were randomly assigned 1:1 to receive REVLIMID® 25 mg orally on days 1-21 of each 28-day cycle and Dexamethasone 40 mg once a week, with or without DARZALEX®. Patients assigned DARZALEX® (D-Rd regimen) received 16 mg/kg weekly for the first 8 weeks (cycles 1 and 2), every other week for 16 weeks (cycles 3 to 6), and then every 4 weeks (cycle 7 and beyond) until disease progression or unacceptable toxicity. Treatment groups were well balanced. The median patient age was 73 years and only 1% of patients were 65 years of age or less whereas 44% of patients were 75 years or older. Cytogenetic risk level could be determined in 642 patients of the total population. Eighty-six percent (86%) of these patients were standard risk and 14% were considered high risk. The Primary end point was Progression Free Survival (PFS). Key Secondary endpoints included Overall Response Rate (ORR), Minimal Residual Disease (MRD) negativity rate (10-5 sensitivity), and Safety.
The prespecified interim analysis occurred with a median follow up of 28 months. DARZALEX® regimen significantly improved PFS with the median PFS not reached with D-Rd compared with 31.9 months in the Rd group (HR=0.55; P<0.0001). This represented a 45% reduction in the risk of progression or death in patients treated with D-Rd. D-Rd also resulted in deeper responses with a Complete Response (CR) or better rate of 47.6% in the D-Rd group compared with 24.7% in the Rd group (P<0.0001). The Very Good Partial Response (VGPR) or better rate was 79.3% in the D-Rd arm compared with 53.1% in the Rd arm (P<0.0001). The MRD-negative rate was more than threefold higher with D-Rd versus Rd at 24% versus 7%, respectively. Higher rates of neutropenia, and leukopenia were observed in the D-Rd arm and the safety profile was consistent with previously reported DARZALEX® studies.
The authors concluded that the addition of DARZALEX®, to REVLIMID® and Dexamethasone significantly reduced the risk of progression or death by 45% in newly diagnosed Multiple Myeloma patients who are transplant-ineligible and these results support D-Rd as a new standard of care for this patient group. Phase 3 Randomized Study of Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) in Patients with Newly Diagnosed Multiple Myeloma (NDMM) Ineligible for Transplant (MAIA). Facon T, Kumar SK, Plesner T, et al. Presented at: ASH Annual Meeting and Exposition; December 4-8, 2018; San Diego, California. Abstract LBA-2.
FDA Approves EMPLICITI® Combination for Relapsed Refractory Multiple Myeloma
SUMMARY: The FDA on November 6, 2018 approved EMPLICITI® (Elotuzumab) in combination with POMALYST® (Pomalidomide) and Dexamethasone for the treatment of adult patients with Multiple Myeloma who have received at least two prior therapies, including REVLIMID® (Lenalidomide) and a Proteasome Inhibitor. Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma (MM) in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Despite the introduction of novel therapies for the treatment of Multiple Myeloma, many patients still face poor outcomes in the Relapsed/Refractory setting.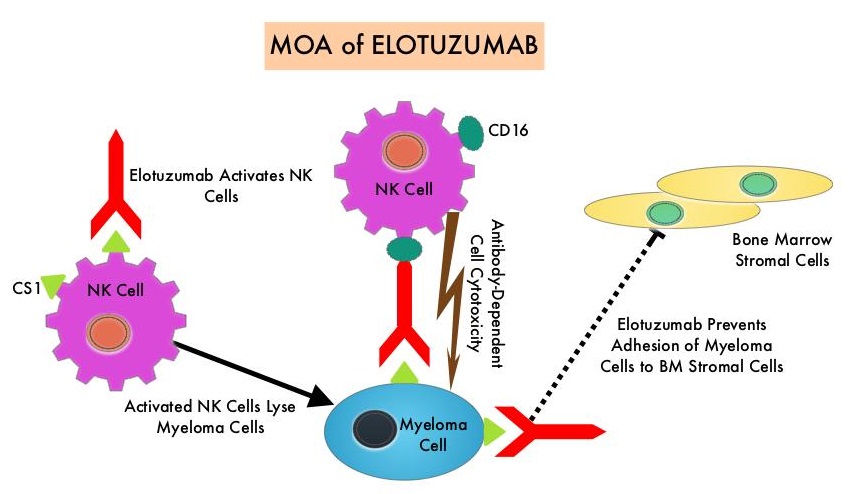
EMPLICITI® is a immunostimulatory monoclonal antibody that binds to the Signal Lymphocyte Activation Molecule – SLAMF7 protein (CS1, CD319), which is highly expressed on Myeloma cells and also expressed on Natural Killer (NK) lymphocytes in the immune system. By virtue of its dual mechanism of action, it targets and destroys Myeloma cells and also enhances the activation of Natural Killer cells. The FDA in November 2015, approved EMPLICITI® for use in combination with REVLIMID® and Dexamethasone for the treatment of patients with Multiple Myeloma who received one to three prior therapies, based on the ELOQUENT-2 trial.
ELOQUENT-3 is a multicenter, randomized, open label, phase II trial in which 117 patients with Relapsed/Refractory Multiple Myeloma were randomly assigned, in a 1:1 ratio, to receive EMPLICITI® plus POMALYST® and Dexamethasone (N=60) – EMPLICITI® group or POMALYST® and Dexamethasone (N= 57) – control group. Treatment was administered in 28-day cycles. Patients in the EMPLICITI® group received EMPLICITI® 10 mg/kg IV days 1, 8, 15, and 22 during cycles 1 and 2 and 20 mg/kg on day 1 of each cycle thereafter. POMALYST® was given at 4 mg orally on days 1 thru 21 of each cycle along with Dexamethasone 40 mg weekly for patients 75 years of age or less or 20 mg for those more than 75 years of age. Treatment was continued until disease progression or unacceptable toxicity. Eligible patients had received 2 or more prior lines of therapy and prophylaxis against thromboembolism was required for all patients. The median age of patients was 67 years. Prior treatments included VELCADE®- Bortezomib (100%), REVLIMID® (99%), KYPROLIS® – Carfilzomib (21%), NINLARO® – Ixazomib (6%), and DARZALEX® – Daratumumab (3%). Close to 55% of patients had undergone Stem Cell transplantation and most patients were refractory to REVLIMID® (87%), a Proteosome Inhibitor (80%), or both (70%). The Primary end point was Progression Free Survival (PFS). The Secondary endpoints included Overall Response Rate (ORR), Complete Response (CR), Stringent Complete Response, Very Good Partial Response and Partial Response Rates.
After a minimum follow up period of 9.1 months, the median PFS was 10.3 months in the EMPLICITI® group and 4.7 months in the control group (HR=0.54; P=0.008), which suggested a 46% reduction in the risk of disease progression. The PFS of EMPLICITI® was consistently observed across all predefined patient subgroups. The Overall Response Rate was 53% in the EMPLICITI® group as compared with 26% in the control group, suggesting a doubling in the Response Rates in the EMPLICITI® group (P=0.0029) with Very Good Partial Responses or better seen in 20% of those in the EMPLICITI® group. The Overall Survival data were immature at the time of the analysis. However, a trend favoring the EMPLICITI® group was observed (HR for death=0.62).
The incidence of serious adverse events was 53% in the EMPLICITI® group and 55% in the control group and the most common treatment-related adverse events in the EMPLICITI® and control groups were neutropenia (18% versus 20%), hyperglycemia (18% versus 11%) and anemia (10% versus 15%), respectively. Adverse events that led to discontinuation of treatment occurred in 18% of the patients in the EMPLICITI® group and in 24% of the patients in the control group.
It was concluded that among patients with Multiple Myeloma who had progressed on REVLIMID® and a Proteosome Inhibitor, EMPLICITI® combined with POMALYST® and Dexamethasone significantly decreased the risk of progression or death and also doubled the Response Rate, compared to POMALYST® plus Dexamethasone alone. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. Dimopoulos MA, Dytfeld D, Grosicki S, et al. N Engl J Med 2018;379:1811-1822
Evaluation of Urine May Aid in the Diagnosis of Lambda Light Chain Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma (MM) in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). Multiple Myeloma evolves from a precursor stage called Monoclonal Gammopathy of Undetermined Significance (MGUS) to MM. Smoldering Multiple Myeloma (SMM) is an intermediate stage in this process of disease evolution.
Monoclonal gammopathies are often diagnosed by electrophoretic examination of the serum and urine proteins. They include Serum Protein ElectroPhoresis (SPEP), Serum Protein Immunofixation electrophoresis (SIFE), Urine Protein ElectroPhoresis (UPEP), Urine Protein Immunofixation electrophoresis (UIFE), and Bone Marrow evaluation. In addition, quantitative assessment of Serum Free Light Chains has been recommended for diagnosis and monitoring of neoplastic monoclonal gammopathies. 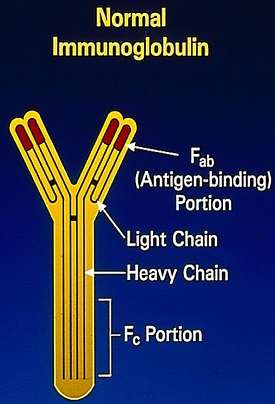
The Y-shaped Immunoglobulins are heterodimeric proteins composed of two heavy (H) and two light (L) chains. The two different types of light chains include Kappa and Lambda, which are distinctive in their amino acid sequence, with normally twice as many Kappa light chains. Malignancy can affect both types of light chains and in Multiple Myeloma. The relevant light chain production can increase, but the increase is more often in the Kappa light chains. Immunoglobulin light chains are produced in excess of the corresponding heavy chains and the excess free light chains can be quantified in serum and can also be detected in the urine, as they, by virtue of their size, are freely filtered through the glomerulus (Bence Jones protein). Excess amounts of free monoclonal light chains in patients with monoclonal gammopathy can produce nephropathy due to precipitation of these proteins in renal tubules.
Serum free Kappa and Lambda light chains are normally present in a ratio of about 0.26 to 1.65 and this ratio is increased in patients with Kappa light chain monoclonal gammopathy and decreased in patients with Lambda chain producing monoclonal gammopathies. Even though alteration in the serum K/L ratio is an important diagnostic criterion for plasma cell neoplasms, there is a high rate of positive results in patients receiving tertiary care, with abnormal K/L ratio noted in 36% of patients and about 90% of these are Kappa light chain dominant.
The serum K/L ratio is however less frequently abnormal and stays normal in patients with Lambda chain lesions even when an abnormal Lambda immunoglobulin is detected in the urine. These variabilities can result in the less-common Lambda chain-associated lesions going undiagnosed. There is a high false negative rate for Lambda dominant K/L ratio in Lambda chain associated monoclonal gammopathy (89% for MGUS, 60% for SMM and 51% for MM). It is estimated that the overall excess false negative K/L ratio rate for Lambda chain lesions, compared to Kappa chain lesions, is approximately 30%. The high false negative rate for the Lambda dominant K/L ratio, in patients with Lambda chain neoplastic monoclonal gammopathies, may be due to under-detection of Lambda light chains, Lambda chains are not produced in as much excess as are Kappa chains resulting in lower rates of Lambda dominant K/L ratio in patient with Lambda light chain neoplastic monoclonal gammopathy, and overproduction of polyclonal Kappa light chains in Lambda chain monoclonal gammopathies, as is usually noted in patients receiving tertiary care.
This study was undertaken by comparing the results of Serum and Urine Protein Electrophoreses with the results of Serum Free Light Chain Assay (SFLCA), to ascertain if the levels of overproduction of the Kappa and Lambda light chain types and their detection rates are different in patients with neoplastic monoclonal gammopathies. The authors performed a retrospective review of SPEP/SIFE, UPEP/UIFE, and SFLCA results from January 2010 through September 2017 from a total of 482 patients. Among these patients, 175 patients had a diagnosis of Neoplastic Monoclonal Gammopathy (MGUS, SMM or MM). , and evaluable results were available to address the questions of this study. Patients with Lymphomas and CLL were excluded.
The authors noted that the serum K/L ratios were appropriately abnormal more often in Kappa light chain disease. In contrast, in those with Lambda light chain disease, the K/L ratios were normal in about 25% of patients but free homogenous Lambda light chains were detectable in urine.
It was concluded that the serum Kappa/Lambda ratio in patients with Lambda light chain disease can be normal in a 25% of patients with Neoplastic Monoclonal Gammopathy and can be missed if not further evaluated with UPEP/UIFE. The authors comment that UPEP/UIFE is under- utilized and the study results question the medical necessity and clinical usefulness of the serum free light chain assay. Serum Free Light Chains in Neoplastic Monoclonal Gammopathies: Relative Under-Detection of Lambda Dominant Kappa/Lambda Ratio, and Underproduction of Free Lambda Light Chains, as Compared to Kappa Light Chains, in Patients With Neoplastic Monoclonal Gammopathies. Lee WS and Singh G. J Clin Med Res. 2018;10:562-569.
Role of Bone-Modifying Agents in Multiple Myeloma American Society of Clinical Oncology Clinical Practice Guideline Update
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). ASCO recently issued a clinical practice guideline update on the role of Bone-Modifying Agents (BMA) in Multiple Myeloma following a systematic literature review of 35 relevant studies by an expert panel. In the updated guidelines, ASCO has recommended expanding the use of bisphosphonates to include all patients being treated for active Multiple Myeloma. The previous guidelines recommended BMAs only for patients with lytic disease. The new recommendation was based on results from the phase III MRC Myeloma IX Trial, which demonstrated the benefit of bisphosphonate therapy in patients with newly diagnosed multiple myeloma who did not have lytic bone disease.
Indications to initiate a BMA – Key Recommendations
Patients with lytic disease on plain radiographs or other imaging studies
For patients with Multiple Myeloma who, on plain radiograph(s) or other imaging studies (MRI or CT scan), have lytic destruction of the bone or compression fracture of the spine from osteopenia, AREDIA® (Pamidronate) 90 mg IV over at least 2 hours or ZOMETA® (Zoledronic acid) 4 mg IV over at least 15 minutes every 3 to 4 weeks is recommended. Alternative treatment includes the use of XGEVA® (Denosumab), a monoclonal antibody that targets Receptor Activator of Nuclear factor Kappa-B Ligand (RANKL).
Patients with solitary plasmacytoma or smoldering (asymptomatic) or indolent myeloma
Starting bisphosphonates in patients with Solitary Plasmacytoma or Smoldering (asymptomatic) or indolent Myeloma is not recommended.
Adjunct to pain control in patients with pain as a result of osteolytic disease and those receiving other interventions for fractures or impending fractures
AREDIA® or ZOMETA® IV is recommended for patients with pain as a result of osteolytic disease and as an adjunctive treatment of patients receiving radiation therapy, analgesics, or surgical intervention to stabilize fractures or impending fractures. XGEVA® is an additional option.
Patients with myeloma with normal plain radiograph or osteopenia in bone mineral density measurements
The Expert Panel supports starting intravenous bisphosphonates in patients with Multiple Myeloma with osteopenia (osteoporosis), but no radiographic evidence of lytic bone disease.
Patients with monoclonal gammopathy of undetermined significance
Starting bisphosphonates in patients with Monoclonal Gammopathy of Undetermined Significance is not recommended, unless osteopenia/osteoporosis exists.
Dosing and selection of BMAs
As a result of increased concerns over renal adverse events, Guidelines recommend that patients with preexisting mild to moderate renal impairment (estimated Creatinine Clearance, 30-60 mL/min) should receive a reduced dosage of ZOMETA®. No changes in infusion time or interval are required. ZOMETA® has not been studied in patients with severe renal impairment and is not recommended for use in these patients. Recent data that compare XGEVA® with ZOMETA® has demonstrated fewer adverse events related to renal toxicity with XGEVA®, and this may be preferred in patients with compromised renal function. AREDIA® 90 mg administered over 4-6 hours is recommended for patients with extensive bone disease and existing severe renal impairment (serum creatinine level more than 3.0 mg/dL (265 µmol/L or an estimated Creatinine Clearance of less than 30 mL/min. Although no dosing guidelines are available for patients with preexisting renal impairment, the Expert Panel recommends that clinicians consider reducing the initial AREDIA® dose in that setting. Infusion times less than 2 hours with AREDIA® or less than 15 minutes with ZOMETA® should be avoided.
Duration of therapy
The Expert Panel suggests that bone-targeting treatment continue for a period of up to 2 years. Less frequent dosing has been evaluated and should be considered in patients with responsive or stable disease. In patients who do not have active Myeloma and are on maintenance therapy, the physician may consider a 3-month interval of bisphosphonate administration. There are no data to support a more precise recommendation for the duration of bisphosphonate therapy in this group of patients. For those patients for whom bisphosphonates were withdrawn after 2 years, the drug should be resumed upon relapse with new-onset Skeletal Related Events. XGEVA® should not be stopped abruptly, given its reversible mechanism of action.
Monitoring
The Expert Panel recommends that serum creatinine should be monitored before each dose of AREDIA® or ZOMETA®, in accordance with FDA-approved labeling. XGEVA® does not require monitoring of renal function. In patients who develop renal deterioration without an apparent cause during bisphosphonate therapy, ZOMETA® or AREDIA® should be withheld. Bisphosphonate therapy can be resumed at the same dosage as that before treatment interruption, when serum creatinine returns to within 10% of the baseline level. XGEVA® requires no dose modification. Serum Calcium should be monitored regularly, and serum Vitamin D levels should be evaluated intermittently. Hypocalcemia is an adverse effect of all bone resorptive agents and is more pronounced with XGEVA®. Patients should be Calcium and Vitamin D repleted. The Expert Panel also recommends intermittent evaluation (every 3-6 months) of all patients receiving AREDIA® or ZOMETA® therapy for the presence of albuminuria, on a spot urine sample. In patients who experience unexplained albuminuria, a 24-hour urine collection should be obtained to assess for more than 500 mg/24 hours of urinary albumin, and discontinuation of the drug is advised until renal problems are resolved. These patients should be reassessed every 3-4 weeks with a 24-hour urine collection for total protein and Urine Protein ElectroPhoresis, and AREDIA® should be reinstituted over a longer infusion time (4 hours or more) and at doses not to exceed 90 mg every 4 weeks, when renal function returns to baseline. The Expert Panel supports the use of screening urinalysis for proteinuria, but underscores that a 24-hour urine collection for the determination of total protein and electrophoresis is required if the test is positive. Although no similar guidelines are available for ZOMETA®, some Expert Panel members recommend that ZOMETA® be reinstituted over a longer infusion time (30 minutes or more).
Biochemical markers
Use of the biochemical markers of bone metabolism to monitor bone-modifying therapy use, is not suggested for routine care.
Osteonecrosis of the jaw
OsteoNecrosis of the Jaw (ONJ) is an uncommon but potentially serious complication of IV bisphosphonates and XGEVA®. The Expert Panel agrees with the recommendations described in the revised FDA label for ZOMETA® and AREDIA®, Dear Doctor letters, a white paper, and various position papers or statements. All patients should receive a comprehensive dental examination and appropriate preventive dentistry before bone-modifying therapy. Active oral infections should be treated, and sites that are at high risk for infection should be eliminated. While on therapy, patients should maintain excellent oral hygiene and avoid invasive dental procedures, if possible. Continuation of a bone-targeting agent in the setting of ONJ has to be individualized and dependent on a risk-benefit ratio and the severity of bone disease.
Role of bone-modifying agents in multiple myeloma: American Society of Clinical Oncology clinical practice guideline update. Anderson K, Ismaila N, Flynn PJ, et al. J Clin Oncol. 2018;36:812-818.
POMALYST® Combination Significantly Improves Progression Free Survival in Relapsed/Refractory Myeloma
SUMMARY: Multiple Myeloma is a clonal disorder of plasma cells in the bone marrow and the American Cancer Society estimates that in the United States, 30,770 new cases will be diagnosed in 2018 and 12,770 patients are expected to die of the disease. Multiple Myeloma in 2018 remains an incurable disease. The therapeutic goal therefore is to improve Progression Free Survival (PFS) and Overall Survival (OS). POMALYST® (Pomalidomide) is a novel, oral, immunomodulatory drug which is far more potent than THALOMID® (Thalidomide) and REVLIMID® (Lenalidomide), and has been shown to be active in REVLIMID® and VELCADE® (Bortezomib) refractory patients. POMALYST® is approved by the FDA for use in combination with Dexamethasone for the treatment of patients with Multiple Myeloma who have received at least 2 prior therapies including REVLIMID® and a Proteasome Inhibitor, and have had disease progression on or within 60 days of completing their last therapy.
POMALYST® has demonstrated synergistic anti-myeloma activity with Dexamethasone and Proteasome Inhibitors. It has been shown to inhibit proliferation of REVLIMID® resistant cells in preclinical studies. With the increasing use of REVLIMID® as first line treatment for patients with Multiple Myeloma, there is a clinically relevant unmet medical need for patients who have progressed on REVLIMID®. The authors herein report the outcomes of a first phase III trial, comparing a combination of POMALYST®, VELCADE® and low dose Dexamethasone (PVd) with VELCADE® and Dexamethasone (Vd), in an entirely post-REVLIMID® treated population.
OPTIMISMM is an international, open label phase III study in which 559 patients with Relapsed/Refractory Multiple Myeloma were randomized in a 1:1 ratio to receive POMALYST® in combination with VELCADE® and low dose Dexamethasone (PVd – N=281) or VELCADE® and Dexamethasone (Vd – N=278). Patients in both treatment groups received VELCADE® 1.3 mg/m² SC, on days 1, 4, 8, and 11 of cycles 1 thru 8, and on days 1 and 8 of cycle 9 and beyond, of each 21 day cycle. Dexamethasone was given to all patients at 20 mg orally daily (10 mg/day if more than 75 years of age) on the days of and after VELCADE® treatment. In the experimental arm, patients received POMALYST® 4 mg orally daily on days 1 thru 14, of each 21 day cycle. The median age was 67.5 years and both treatment groups were well balanced. All patients had prior treatment with REVLIMID® and 70% were refractory to this agent, whereas 72% of the patients had prior treatment with VELCADE® and 68% were refractory to the last treatment. The median number of prior treatment lines was 2 and approximately 40% of the patients in both treatment groups had one prior line of therapy. The percentage of patients with high-risk cytogenetics such as del(17p), t(4;14), and or t(14;16]), was similar in both treatment groups. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Overall Response Rate (ORR), Duration of Response, and Safety.
At a median follow up of 16 months, the median PFS was 11.2 months with PVd compared with 7.1 months with Vd alone (HR=0.61; P<0.0001). This meant a 39% reduction in the risk of progression or death with POMALYST®, VELCADE® and low dose Dexamethasone combination, compared with VELCADE® and low dose Dexamethasone alone. This PFS benefit was noted regardless of age, performance status, high-risk cytogenetics, number of prior therapies, and types of prior therapy. The OS data are not mature. The most common side effects of the drug combinations were neutropenia, infections, and thrombocytopenia, which were manageable.
It was concluded that in the treatment of Multiple Myeloma, there remains an unmet medical need for those patients who have received REVLIMID® based therapies and are in early relapse. OPTIMISMM is the only phase III study to date in early Relapsed/Refractory Multiple Myeloma, that has demonstrated a significant and clinically meaningful PFS improvement in patients who have previously received REVLIMID® and especially those who are refractory to REVLIMID®, suggesting that the combination of POMALYST®, VELCADE® and low dose Dexamethasone may be a new standard of care in patients with Relapsed/Refractory Multiple Myeloma, with prior exposure to REVLIMID®. Pomalidomide (POM), bortezomib, and lowâ€dose dexamethasone (PVd) vs bortezomib and low-dose dexamethasone (Vd) in lenalidomide (LEN)-exposed patients (pts) with relapsed or refractory multiple myeloma (RRMM): Phase 3 OPTIMISMM trial. Richardson PG, Rocafiguera AO, Beksac M, et al. J Clin Oncol 36, 2018 (suppl; abstr 8001)